pygidSIM calculates GIWAXS patterns from CIF files or other crystal structure descriptions.
![]()
pip install pygidsimFirst, clone the repository:
git clone https://github.com/mlgid-project/pygidSIM.gitThen, to install all required modules, navigate to the cloned directory and execute:
cd pygidSIM
pip install -e .For development and testing, install with development dependencies:
pip install -e .[dev]The project uses pytest for testing. To run the test suite:
# Run all tests
pytest
# Run tests with coverage report
pytest --cov=pygidsim --cov-report=html
# Run tests in parallel
pytest -n autoTo calculate the peak positions and their intensities in 2D GIWAXS pattern (qxy, qz) from a CIF file with the default orientation hkl = [001] (vector normal to the substrate, i.e. {001} contact plane) run the following:
from pygidsim.experiment import ExpParameters
from pygidsim.giwaxs_sim import GIWAXSFromCif
params = ExpParameters(
q_xy_range=(0, 2.7),
q_z_range=(0, 2.7),
en=18000
) # experimental parameters
el = GIWAXSFromCif(path_to_cif, params)
q_2d, intensity = el.giwaxs.giwaxs_sim() # q_2d is array with shape (2, peaks number)To add a crystal rotation use the argument orientation with the value "random" or a numpy array containing the
corresponding Miller indices [hkl]:
q_2d, intensity = el.giwaxs.giwaxs_sim(orientation='random')
q_2d, intensity = el.giwaxs.giwaxs_sim(orientation=numpy.array([2., 0., 1.]))For 3D powder diffraction simulation (non-oriented case) use orientation=None:
q_1d, intensity_1d = el.giwaxs.giwaxs_sim(orientation=None)To return the Miller indices, you can use the argument return_mi = True:
q_2d, intensity, mi = el.giwaxs.giwaxs_sim(return_mi=True)To restrict the maximum Miller index for simulation use the argument max_mi:
q_2d, intensity = el.giwaxs.giwaxs_sim(max_mi=3)To calculate a GIWAXS pattern from your own description, use the following example:
import numpy as np
from pygidsim.giwaxs_sim import GIWAXS, Crystal
# space group number
spgr = 221 # alternatively, use e.g. '146:R'
# lattice parameters [a, b, c, α, β, γ]
lat_par = np.array([6.3026, 6.3026, 6.3026, 90., 90., 90.], dtype=np.float32)
# list of atoms
atoms = np.array(['Pb', 'I', 'I', 'I', 'N'])
# relative atom positions
atom_positions = np.array(
[[0., 0., 0.],
[0.5, 0., 0.],
[0., 0.5, 0.],
[0., 0., 0.5],
[0.5, 0.5, 0.5]], dtype=np.float32
)
# occupancies of the corresponding sites
occupancy = np.array([1., 1., 1., 1., 1.], dtype=np.float32)
cr = Crystal(lat_par, spgr, atoms, atom_positions, occupancy)
el = GIWAXS(cr, params)
q_2d, intensity = el.giwaxs_sim(orientation='random')The intensities are set to one in case the arguments atoms or/and atom_positions are not provided.
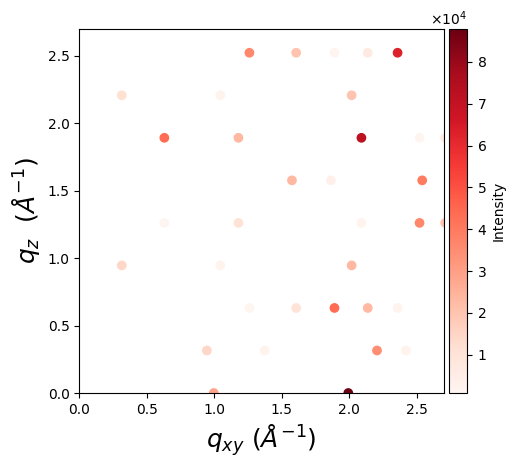
One can visualize a GIWAXS pattern using matplotlib:
import matplotlib.pyplot as plt
from mpl_toolkits.axes_grid1.axes_divider import make_axes_locatable
fig, ax = plt.subplots(figsize=(5, 5))
ax.set_aspect('equal')
scatter = ax.scatter(*q_2d, c=intensity, cmap='Reds')
ax.set_xlabel(r'$q_{xy}$ $(Å^{-1})$', fontsize=18)
ax.set_ylabel(r'$q_{z}$ $(Å^{-1})$', fontsize=18)
divider = make_axes_locatable(ax)
# Append a new axes for the color bar to the right of the current axes
cax = divider.append_axes("right", size="5%", pad=0.05)
# Create color bar in the new axes
colorbar = fig.colorbar(scatter, cax=cax)
colorbar.set_label('Intensity')
plt.show()
If you use this package in your research, please cite it as follows:
Romodin, M., Starostin, V., Lapkin, D., Hinderhofer, A., & Schreiber, F. (2025).
mlgid-project/pygidSIM: v0.1.1. Zenodo. https://doi.org/10.5281/zenodo.17609569