diff --git a/docs/_includes/head_custom.html b/docs/_includes/head_custom.html
new file mode 100644
index 0000000..d57da84
--- /dev/null
+++ b/docs/_includes/head_custom.html
@@ -0,0 +1,14 @@
+
+
+
+
+
+
+
+
diff --git a/docs/courses/CSRC cluster/PBS_torque.md b/docs/courses/CSRC cluster/PBS_torque.md
new file mode 100644
index 0000000..3f1b360
--- /dev/null
+++ b/docs/courses/CSRC cluster/PBS_torque.md
@@ -0,0 +1,76 @@
+---
+layout: default
+title: Job Submission on Cinci
+nav_order: 1
+parent: CSRC cluster
+grand_parent: Lab Documentation
+---
+
+# Job Submission on Cinci
+
+The Cinci server used at the Luque Lab uses PBS (Portable Batch System) Torque which is a resource management system for submitting and controlling jobs on supercomputers, clusters and grids.
+
+The Cinci server has 20 compute nodes - node 1 through node 20 amongst which 4 are higher performance/memorgy compute nodes - node 17 through node 20. Nodes 1 through 16 have 16 processors each and nodes 17 through 20 have 24 processors each.
+
+Job submission is accomplished using the qsub command. The PBS command file is specified as a filename on the qsub command line. Once we submit a script via qsub which is then sent to the scheduling system. The scheduler will then find free resources and run the script on an available node.
+
+The resource requests can be made using either the script or command line arguments.
+
+**Using command line**:
+
+
+```bash
+qsub -l nodes=16
+```
+ Specifies that we are requesting 16 nodes of any type. If the number of nodes are not specified 1 node is taken.
+
+
+```bash
+qsub -l nodes=16:ppn=24
+```
+Specifies that we are requesting 24 processors on each of the 16 nodes
+
+```bash
+qsub -l nodes=1:ppn=1,pmem=100MB,walltime=5:00
+```
+Specifies that we are requestion one node, one processor on that node, 100 mega bytes per process and a walltime of five minutes.
+
+**Using scripts**:
+
+```bash
+#PBS -N Name
+#PBS -l nodes=1:ppn=1,walltime=01:00
+
+echo "Hello World"
+```
+Here we are giving a job name, specifying that we need one processor on 1 node for 1 minute.
+
+
+A list of other arguments and commands can be found [here](https://www.cqu.edu.au/eresearch/high-performance-computing/hpc-user-guides-and-faqs/pbs-commands).
+
+**Monitoring Jobs**
+
+The qstat command is used to monitor submitted jobs.
+
+```bash
+qstat -f
+```
+Specifies that a full status display be written to standard out. The [time] value is the amount of walltime, in seconds, remaining for the job.
+
+More qstat options can be found [here](http://docs.adaptivecomputing.com/torque/3-0-5/commands/qstat.php).
+
+
+**Canceling Jobs**
+
+Submitted jobs can be cancelled using the qdel command. The only parameter is the ID of the job to be canceled. For example if we want to kill the job with an ID 314
+
+```bash
+qdel 314
+```
+
+To delete all the the jobs
+```bash
+qdel all
+```
+
+More qdel options can be found [here](http://docs.adaptivecomputing.com/torque/3-0-5/commands/qdel.php).
diff --git a/docs/courses/CSRC cluster/anaconda_environment.md b/docs/courses/CSRC cluster/anaconda_environment.md
new file mode 100644
index 0000000..36e86a1
--- /dev/null
+++ b/docs/courses/CSRC cluster/anaconda_environment.md
@@ -0,0 +1,50 @@
+---
+layout: default
+title: Entering the conda environment on cinci cluster
+nav_order: 2
+parent: CSRC cluster
+grand_parent: Lab Documentation
+---
+
+# Entering the conda environment on cinci cluster
+
+~~~
+# .bashrc
+# User specific aliases and functions
+alias rm='rm -i'
+alias cp='cp -i'
+alias mv='mv -i'
+~~~
+
+.bashrc initializes an interactive shell session. The bottom three lines are to prompt the user before deletion, overwriting and moving the file.
+
+~~~
+# Source global definitions
+if [ -f /etc/bashrc ]; then
+ . /etc/bashrc
+fi
+
+# added by Anaconda3 5.3.0 installer
+# >>> conda init >>>
+# !! Contents within this block are managed by 'conda init' !!
+__conda_setup="$(CONDA_REPORT_ERRORS=false '/usr/local/anaconda3/bin/conda' shell.bash hook 2> /dev/null)"
+if [ $? -eq 0 ]; then
+ \eval "$__conda_setup"
+else
+ if [ -f "/usr/local/anaconda3/etc/profile.d/conda.sh" ]; then
+ . "/usr/local/anaconda3/etc/profile.d/conda.sh"
+ CONDA_CHANGEPS1=false conda activate base
+ else
+ \export PATH="/usr/local/anaconda3/bin:$PATH"
+ fi
+fi
+unset __conda_setup
+# <<< conda init <<<
+~~~
+
+/etc/bashrc contains system wide funtions and aliases. Environment related files and programs are available in etc/profile.
+The if .. fi statement is the fundamental control statement that allows shell to make decisions and execute statements conditionally.
+
+
+
+
diff --git a/docs/courses/CSRC cluster/bashrc-ana3.bashrc b/docs/courses/CSRC cluster/bashrc-ana3.bashrc
new file mode 100644
index 0000000..b0bf1b2
--- /dev/null
+++ b/docs/courses/CSRC cluster/bashrc-ana3.bashrc
@@ -0,0 +1,28 @@
+# .bashrc
+
+# User specific aliases and functions
+
+alias rm='rm -i'
+alias cp='cp -i'
+alias mv='mv -i'
+
+# Source global definitions
+if [ -f /etc/bashrc ]; then
+ . /etc/bashrc
+fi
+# added by Anaconda3 5.3.0 installer
+# >>> conda init >>>
+# !! Contents within this block are managed by 'conda init' !!
+__conda_setup="$(CONDA_REPORT_ERRORS=false '/usr/local/anaconda3/bin/conda' shell.bash hook 2> /dev/null)"
+if [ $? -eq 0 ]; then
+ \eval "$__conda_setup"
+else
+ if [ -f "/usr/local/anaconda3/etc/profile.d/conda.sh" ]; then
+ . "/usr/local/anaconda3/etc/profile.d/conda.sh"
+ CONDA_CHANGEPS1=false conda activate base
+ else
+ \export PATH="/usr/local/anaconda3/bin:$PATH"
+ fi
+fi
+unset __conda_setup
+# <<< conda init <<<
diff --git a/docs/courses/CSRC cluster/checkv.md b/docs/courses/CSRC cluster/checkv.md
new file mode 100644
index 0000000..8782f00
--- /dev/null
+++ b/docs/courses/CSRC cluster/checkv.md
@@ -0,0 +1,52 @@
+---
+layout: default
+title: Submitting jobs on cinci cluster - CheckV
+nav_order: 3
+parent: CSRC cluster
+grand_parent: Lab Documentation
+---
+
+# Using qsub to submit bash scripts to run CheckV
+
+The official README for CheckV can be found [here](https://bitbucket.org/berkeleylab/checkv/src/master/).
+
+For this code to run we are assuming that you have bashrc-ana3.bashrc in your home directory.
+
+~~~
+#!/bin/bash
+#PBS -l nodes=2:ppn=24
+#PBS -l walltime=02:00:00
+~~~
+
+The first line us the most common shell used as default shel for user login of the linux system. It is used to instruct the operating system to use bash as a command interpreter. The second ans third lines request resources for the job. This job requests two nodes with 24 ppn (processors per node). This combination would run the job using two of the 12 core 24 thread CPUs on the cinci cluster.
+
+~~~
+cat ${PBS_NODEFILE}
+~~~
+The nodefile contains a list of all nodes the job has allocated with an entry for every CPU.
+
+~~~
+cd
+source bashrc-ana3
+checkv end_to_end input_file.fna output_directory -d location_of_the_databse
+~~~
+The first line is the command which is used to change the current working directory in Linux. The second line is to enter the anaconda environment. The third line is to run CheckV using a single command to run the fill pipeline
+
+Here is an example of how we can do it.
+~~~
+checkv end_to_end ~/Test_CheckV/Test_sequence.fna ~/Test_CheckV/result -d /usr/local/checkv-db-v1.1
+~~~
+
+Another way to run CheckV using individual commands for each step in the pipeline
+~~~
+checkv contamination input_file.fna output_directory
+checkv completeness input_file.fna output_directory
+checkv complete_genomes input_file.fna output_directory
+checkv quality_summary input_file.fna output_directory
+~~~
+
+You can now submit your script as a job using the command:
+~~~
+qsub checkv.sh
+~~~
+
diff --git a/docs/courses/CSRC cluster/checkv.sh b/docs/courses/CSRC cluster/checkv.sh
new file mode 100644
index 0000000..91a4976
--- /dev/null
+++ b/docs/courses/CSRC cluster/checkv.sh
@@ -0,0 +1,15 @@
+#!/bin/bash
+#PBS -l nodes=2:ppn=24
+#PBS -l walltime=02:00:00
+
+echo "Running on: "
+cat ${PBS_NODEFILE} # The nodefile contains a list of all nodes the job has allocated with an entry for every CPU
+
+echo
+echo "Program Output begins: "
+
+cd # going into the home directory
+source bashrc-ana3 # entering the anaconda environment
+checkv end_to_end ~/Test_CheckV/Test_sequence.fna ~/Test_CheckV/result -d /usr/local/checkv-db-v1.1
+
+# checkv end_to_end input_file.fna output_directory -d location_of_the_databse
diff --git a/docs/courses/CSRC cluster/csrc_cluster.md b/docs/courses/CSRC cluster/csrc_cluster.md
new file mode 100644
index 0000000..c7ef181
--- /dev/null
+++ b/docs/courses/CSRC cluster/csrc_cluster.md
@@ -0,0 +1,11 @@
+---
+layout: default
+title: CSRC cluster
+nav_order: 4
+parent: Lab Documentation
+has_children: true
+---
+
+## CSRC cluster
+
+This website offers comprehensive tutorials on efficiently running programs on the CSRC Cinci Cluster, a powerful multicore CPU cluster.
diff --git a/docs/courses/CSRC cluster/okular.md b/docs/courses/CSRC cluster/okular.md
new file mode 100644
index 0000000..169d428
--- /dev/null
+++ b/docs/courses/CSRC cluster/okular.md
@@ -0,0 +1,52 @@
+---
+layout: default
+title: Viewing PDF's on cinci
+nav_order: 5
+parent: CSRC cluster
+grand_parent: Lab Documentation
+---
+
+# Viewing PDF's on the cinci cluster
+
+The cinci cluster uses okular as the document viewer. Following are the steps that need to be followed to use ssh with x11 forwarding be able to access okular.
+
+
+## Accessing okular using Linux/ Mac
+1. Download and install XQuartz on macOS
+ * XQuartz package can be found [here](https://www.xquartz.org/)
+ * Once this package is downloaded, install the server and follow the instructions provided on the dialogue box to complete the installation process
+ * XQuartz can also be downloaded using the brew command on the terminal
+
+ ``` brew install -cask xquartz```
+
+2. Reboot your mac
+ * Click on the Apple icon and then restart or use the following command on the terminal
+
+ ``` sudo reboot ```
+
+3. Log into cinci using the SSH command with -X or -Y flages
+
+ ```ssh -X user@cinci.sdsu.edu``` or
+
+ ```ssh -Y user@cinci.sdsu.edu```
+4. Open required file using the following command
+
+ ``` okular path/doc.pdf ```
+
+5. More command line options for okular can be found [here](https://docs.kde.org/stable5/en/okular/okular/command-line-options.html)
+
+
+
+## Accessing okular using Windows
+1. Download and install PuTTY from [here](https://www.chiark.greenend.org.uk/~sgtatham/putty/latest.html)
+2. Create a session under the 'Session' category. Enter 'cinci.sdsu.edu' under 'Host Name' and under 'Saved Sessions'
+3. Select SSH as the Connection type
+4. Click on the 'plus' of the SSH category, select X11, and check 'Enable X11 forwarding'
+5. Select the 'Session' category, and clieck on 'Save'.
+6. Conntect to cinci.sdsu.edu by clicking 'Open'.
+7. Log in using your username and password.
+8. Open required file using the following command
+
+ ``` okular path/doc.pdf ```
+
+9. More command line options for okular can be found [here](https://docs.kde.org/stable5/en/okular/okular/command-line-options.html).
diff --git a/docs/courses/CSRC cluster/pyCapsid.md b/docs/courses/CSRC cluster/pyCapsid.md
new file mode 100644
index 0000000..07b964f
--- /dev/null
+++ b/docs/courses/CSRC cluster/pyCapsid.md
@@ -0,0 +1,30 @@
+---
+layout: default
+title: Job Submission on Cinci - pyCapsid
+nav_order: 6
+parent: CSRC cluster
+grand_parent: Lab Documentation
+---
+
+1. The first step to running pyCapsid on the cluster is installing pyCapsid in dedicated conda environment as described in
+its [installation documentation.](https://luquelab.github.io/pyCapsid/installation/#via-conda)
+2. Once you've done that, activate the environment using `conda activate` and note the path to the python interpreter of that environment using
+`type python`. For example:
+
+3. Create a folder in which you will store the results of pyCapsid. Note the path of the folder using `pwd`.
+4. Inside that folder create a file named `config.toml` or download an example config.toml from [here](https://github.com/luquelab/pyCapsid/blob/main/docs/tutorial/config_simple.toml).
+Adjust the contents of this config file to the desired set of parameters. A more detailed config file with more of the available options can be found [here](https://github.com/luquelab/pyCapsid/blob/main/docs/tutorial/conf_example.toml).
+5. Next create a python file with your preferred name, i.e. `run_pycap.py`. The contents of that file should look like this:
+ ```python
+ #!/usr/dataB/luquelab/members/ctbrown/miniconda3/envs/pycapsid/bin/python # This is the path to your python interpreter noted in step 2
+ #PBS -l nodes=1:ppn=24 # This requests a single node with 24 processors per node. This corresponds to the 4 higher quality nodes on the CSRC cluster. Remove the ppn requirement to use any node.
+ #PBS -l walltime=18:00:00 # This specifies the maximum time the job will run before being terminated
+ #PBS -d "/usr/dataB/luquelab/members/ctbrown/pyCapsid/pyCapsid/run/" # This specifies the working directory, and should be the directory you created in step 3
+ #PBS -j oe # This combines the standard output and standard error logs
+
+ from pyCapsid import run_capsid_report
+ run_capsid_report('config.toml') # make sure the filename provided here is the same as the config file you created.
+ ```
+6. Check that you set the parameters in `config.toml` correctly and submit the job using `qsub run_pycap.py`.
+7. This will create a folder with the same name that was specified in the config file. You can copy this folder to your
+local machine using the scp command.
diff --git a/docs/courses/CSRC cluster/python_path_example.png b/docs/courses/CSRC cluster/python_path_example.png
new file mode 100644
index 0000000..0c2c005
Binary files /dev/null and b/docs/courses/CSRC cluster/python_path_example.png differ
diff --git a/docs/courses/CSRC cluster/vibrant.md b/docs/courses/CSRC cluster/vibrant.md
new file mode 100644
index 0000000..507d37c
--- /dev/null
+++ b/docs/courses/CSRC cluster/vibrant.md
@@ -0,0 +1,46 @@
+---
+layout: default
+title: Submitting jobs on cinci cluster - VIBRANT
+nav_order: 4
+parent: CSRC cluster
+grand_parent: Lab Documentation
+---
+
+# Using qsub to submit bash scripts to run VIBRANT
+
+The official README for CheckV can be found [here](https://github.com/AnantharamanLab/VIBRANT).
+
+For this code to run we are assuming that you have bashrc-ana3.bashrc in your home directory.
+
+~~~
+#!/bin/bash
+#PBS -l nodes=2:ppn=24
+#PBS -l walltime=02:00:00
+~~~
+
+The first line us the most common shell used as default shel for user login of the linux system. It is used to instruct the operating system to use bash as a command interpreter. The second ans third lines request resources for the job. This job requests two nodes with 24 ppn (processors per node). This combination would run the job using two of the 12 core 24 thread CPUs on the cinci cluster.
+
+~~~
+cat ${PBS_NODEFILE}
+~~~
+The nodefile contains a list of all nodes the job has allocated with an entry for every CPU.
+
+~~~
+cd
+source bashrc-ana3
+cd ../../usr/local/VIBRANT-1.2.1/
+python3 VIBRANT_run.py -i input_file.fna -folder output_directory
+~~~
+The first line is the command which is used to change the current working directory in Linux. The second line is to enter the anaconda environment. The third line is to go to the directory where VIBRANT is installed. The fourth line is to run VIBRANT using python3.
+
+Here is an example
+~~~
+python3 VIBRANT_run.py -i ~/Test_VIBRANT/DTRs_ctgs_1-10.fna -folder ~/Test_VIBRANT/Test_results
+~~~
+
+you can now submit your script as a job using the command:
+
+~~~
+qsub vibrant.sh
+~~~
+
diff --git a/docs/courses/CSRC cluster/vibrant.sh b/docs/courses/CSRC cluster/vibrant.sh
new file mode 100644
index 0000000..95d6499
--- /dev/null
+++ b/docs/courses/CSRC cluster/vibrant.sh
@@ -0,0 +1,30 @@
+#!/bin/bash
+#PBS -l nodes=1:ppn=24
+#PBS -l walltime=02:00:00
+
+cd ~/Test_VIBRANT # cd into the directory with the test file
+echo
+echo "Program Output begins: "
+
+head -10 DTRs_20kb.fna | tail -10 > DTRs_ctgs_1-10.fna;
+head -20 DTRs_20kb.fna | tail -10 > DTRs_ctgs_11-20.fna;
+head -30 DTRs_20kb.fna | tail -10 > DTRs_ctgs_21-30.fna;
+head -40 DTRs_20kb.fna | tail -10 > DTRs_ctgs_31-40.fna;
+head -50 DTRs_20kb.fna | tail -50 > DTRs_ctgs_41-50.fna;
+
+
+cd # going into the home directory
+source bashrc-ana3 # entering the anaconda environment
+
+cd ../../usr/local/VIBRANT-1.2.1/
+
+echo "Running VIBRANT"
+
+python3 VIBRANT_run.py -i ~/Test_VIBRANT/DTRs_ctgs_1-10.fna -folder ~/Test_VIBRANT/Test_results
+python3 VIBRANT_run.py -i ~/Test_VIBRANT/DTRs_ctgs_11-20.fna -folder ~/Test_VIBRANT/Test_results
+python3 VIBRANT_run.py -i ~/Test_VIBRANT/DTRs_ctgs_21-30.fna -folder ~/Test_VIBRANT/Test_results
+python3 VIBRANT_run.py -i ~/Test_VIBRANT/DTRs_ctgs_31-40.fna -folder ~/Test_VIBRANT/Test_results
+python3 VIBRANT_run.py -i ~/Test_VIBRANT/DTRs_ctgs_41-50.fna -folder ~/Test_VIBRANT/Test_results
+
+
+
diff --git a/docs/courses/GitHub/GitHub_LuqueLab.md b/docs/courses/GitHub/GitHub_LuqueLab.md
new file mode 100644
index 0000000..a9992f9
--- /dev/null
+++ b/docs/courses/GitHub/GitHub_LuqueLab.md
@@ -0,0 +1,28 @@
+---
+layout: default
+title: GitHub for Luque Lab
+nav_order: 2
+parent: Lab Documentation
+has_children: true
+---
+
+# **GitHub fundamentals for lab members**
+
+[Luque Lab](https://www.luquelab.com/team.html) organizes all the projects on [Luque Lab Github organization](https://github.com/luquelab). Below are the fundamentals of using Git and Github. Git and Github are tools generally used by software developers and creative coders. [Git](https://git-scm.com/downloads) is an actual version control software application which can be run anywhere. [GitHub]((https://github.com/)) is a web service for software development and version control using git. One needs to signup or have an account to access.
+
+## External Tutorials
+- [Git and Github for beginners - Crash Course](https://www.youtube.com/watch?v=RGOj5yH7evk&ab_channel=freeCodeCamp.org)
+- [Git command line fundamentals](https://www.youtube.com/watch?v=HVsySz-h9r4)
+- [Git and Github playlist](https://www.youtube.com/watch?v=3RjQznt-8kE&list=PL4cUxeGkcC9goXbgTDQ0n_4TBzOO0ocPR)
+
+## Lab Tutorials
+- [GitHub Repositories](https://luquelab.github.io/Athena/courses/GitHub/GitHub_repos.html)
+- [Accessing Git though command line](https://luquelab.github.io/Athena/courses/GitHub/Git_commandline.html)
+- [Creating GitHub pages with Jekyll](https://luquelab.github.io/Athena/courses/GitHub/GitHub_pagesJekyll.html)
+- [Markdown Tutorials](https://luquelab.github.io/Athena/courses/markdown_tutorials/)
+- [Adding Dropdown Menus for GitHub pages](https://luquelab.github.io/Athena/courses/markdown_tutorials/add_dropdowns.html)
+- [Adding Images to Markdown Files](https://luquelab.github.io/Athena/courses/markdown_tutorials/add_images.html)
+- [GitHub Backup](https://luquelab.github.io/Athena/courses/GitHub/backup_github.html)
+
+
+
diff --git a/docs/courses/GitHub/GitHub_pagesJekyll.md b/docs/courses/GitHub/GitHub_pagesJekyll.md
new file mode 100644
index 0000000..f47d758
--- /dev/null
+++ b/docs/courses/GitHub/GitHub_pagesJekyll.md
@@ -0,0 +1,19 @@
+---
+layout: default
+title: GitHub pages with Jekyll
+nav_order: 3
+parent: GitHub for Luque Lab
+grand_parent: Lab Documentation
+---
+
+# **GitHub pages with Jekyll**
+- Jekyll takes Markdown and HTML files to create static site with built-in support for GitHub Pages.
+- Jekyll settings can be configured by editing the *_config.yml* file.
+- To add title, layout, order and drop-downs you can add YAML from matter at the beginning to any Markdown file.
+
+
+Here are some useful resources for setting up a Github Pages site with Jekyll
+- [Getting Started with Github Pages](https://www.youtube.com/watch?v=QyFcl_Fba-k&ab_channel=TheNetNinja)
+- [About GitHub Pages and Jekyll](https://docs.github.com/en/pages/setting-up-a-github-pages-site-with-jekyll/about-github-pages-and-jekyll)
+- [Creating a GitHub Pages site with Jekyll](https://docs.github.com/en/pages/setting-up-a-github-pages-site-with-jekyll/creating-a-github-pages-site-with-jekyll)
+- [Adding content](https://docs.github.com/en/pages/setting-up-a-github-pages-site-with-jekyll/adding-content-to-your-github-pages-site-using-jekyll)
diff --git a/docs/courses/GitHub/GitHub_repos.md b/docs/courses/GitHub/GitHub_repos.md
new file mode 100644
index 0000000..499ee57
--- /dev/null
+++ b/docs/courses/GitHub/GitHub_repos.md
@@ -0,0 +1,24 @@
+---
+layout: default
+title: GitHub Repos
+nav_order: 1
+parent: GitHub for Luque Lab
+grand_parent: Lab Documentation
+---
+
+# **GitHub Repositories**
+- Repository/ Repo is another word for a project and can have multiple files in it.
+- Go through this tutorial to familiarize yourself with repositories: [Tutorial](https://docs.github.com/en/get-started/quickstart/create-a-repo)
+
+## Keywords
+- Commit
+ - A commit is an individual change to a file similar to saving a file. Each commit can have a note/ message with it and allows you to keep a record of all the changes made until that moment. Each commit has it's own commit hash which is the unique identifier for that particular commit.
+
+- Branch
+ - Branches allow you to experiment with your project without changing the main project. Once you edit a file in your branch it won't be uptaded in your main master branch until you create a pull request and merge the branch.
+
+- Pull requests and merge
+ - If you decide to keep the changes made in the branch into the main branch you send a oull request to merge the current branch. Once the request is accepted the branch is merged into the master.
+
+- Forks and Pull requests
+ - Forking is making a copy of a repository or an instance which is not yours and having it under your account without affecting the original version. You send in a pull request if you want to contribute your changes back to the original. It is up to the main account holder to accept or reject the pull request.
diff --git a/docs/courses/GitHub/Git_commandline.md b/docs/courses/GitHub/Git_commandline.md
new file mode 100644
index 0000000..9418979
--- /dev/null
+++ b/docs/courses/GitHub/Git_commandline.md
@@ -0,0 +1,44 @@
+---
+layout: default
+title: Git though command line
+nav_order: 2
+parent: GitHub for Luque Lab
+grand_parent: Lab Documentation
+---
+
+# **Git through command line**
+
+Here are some useful resources about using git through command line:
+- [Start using Git on the command line](https://docs.gitlab.com/ee/gitlab-basics/start-using-git.html)
+- [Git Cheat Sheet](https://www.atlassian.com/git/tutorials/atlassian-git-cheatsheet)
+- [Git command-line fundamentals](https://www.youtube.com/watch?v=HVsySz-h9r4)
+
+## Basic git commands
+- git --version
+ - To check the version of git
+
+- git init
+ - This command initializes your local repository
+
+- git status
+ - To check the status of the repository.
+ - Working area: Files that are not yet added to the repository are stored here (Untracked files)
+ - Staging area: Files that are going to be a part of the next commit
+
+- git add
+ - To add files into the staging area from your workspace
+
+- git commit -m "your message"
+ - This command lets you add your files to the local repository, -m gives you the option to pass a message
+
+- git log
+ - This commands allows you to view all the commits and changes made by the user
+
+- git clone "Repo_url"
+ - This command makes a copy of the remote repository and it's branches on your local computer
+
+- git branch branch_name
+ - This command creates a new branch
+
+- git checkout branch_name
+ - This switches you from one branch to another
diff --git a/docs/courses/GitHub/backup_github.md b/docs/courses/GitHub/backup_github.md
new file mode 100644
index 0000000..2c88094
--- /dev/null
+++ b/docs/courses/GitHub/backup_github.md
@@ -0,0 +1,53 @@
+---
+layout: default
+title: Lab GitHub Backup
+nav_order: 7
+parent: GitHub for Luque Lab
+grand_parent: Lab Documentation
+---
+# Manual Github Backup
+
+The [Luque lab](https://www.luquelab.com/team.html) follows the 3-2-1 backup strategy for backing up all lab-related data. This strategy states that you should have 3 copies of your data, on 2 different media, with 1 copy being off site. All the repositories on the [Luque Lab GitHub organization](https://github.com/luquelab) are backed up frequently using two bash scripts.
+
+The first script clones a repository that already exists on GitHub, including all of the files, branches, and commits. This script is run when new repositories are created on the organization's website or the names of the repositories are changed. Each repository is cloned using the git clone command. Here is an example of cloning the Athena repository from the Luque Lab GitHub organization.
+
+```
+ git clone https://github.com/luquelab/Athena.git;
+```
+
+The second script fetches and pulls each repository. This is to make sure each repository on your device is up to date with the respoitories on Github. For this following code to run without warnings or errors, the subfolders in the main backup folder should not be changed or include any other folders not available as repositories on GitHub. This is because the code goes through every subfolder and fetches the repository using the metadata available on your device in the particular subfolder. The code is as follows.
+
+```
+#!/bin/bash
+for dir in */; do
+ echo "$dir"
+
+ echo "Entering directory";
+ cd $dir;
+
+ echo "Fetch";
+ git fetch --all;
+
+ echo "Pull";
+ git pull --all;
+
+ echo "All the branches";
+ git branch -a
+
+ echo "Exiting directory";
+ cd ..;
+done
+```
+
+The backup process is scheduled to run weekly. Upon completing the GitHub backup, the entire luquelab folder from the Cinci server is backed up to the Notos server using the following command:
+
+```
+ rsync -av --delete --force luquelab agarwal@notos.sdsu.edu:/mnt/beegfs/home/luquelab/backup
+```
+
+This ensures a seamless and secure transfer of data, maintaining the integrity of the luquelab folder for future reference and restoration purposes.
+
+
+
+
+
diff --git a/docs/courses/GitHub/github_website.md b/docs/courses/GitHub/github_website.md
new file mode 100644
index 0000000..64bae23
--- /dev/null
+++ b/docs/courses/GitHub/github_website.md
@@ -0,0 +1,10 @@
+---
+layout: default
+title: GitHub Lab Website Backup
+nav_order: 7
+parent: GitHub for Luque Lab
+grand_parent: Lab Documentation
+---
+# GitHub to official website
+
+The lab website is available in two versions. The GitHub Pages version can be accessed at [https://luquelab.github.io/website/](https://luquelab.github.io/website/) and it contains the latest lab accomplishments. The main website, [https://www.luquelab.com/](https://www.luquelab.com/), is built using a third-party website generator called Weebly. Both versions provide valuable information about the lab and its activities.
diff --git a/docs/courses/Meetingowl.md b/docs/courses/Meetingowl.md
new file mode 100644
index 0000000..1abd940
--- /dev/null
+++ b/docs/courses/Meetingowl.md
@@ -0,0 +1,19 @@
+---
+layout: default
+title: Meeting Owl
+nav_order: 10
+parent: Lab Documentation
+---
+
+# Setting Up MeetingOwl Pro
+The Luque lab uses MeetingOwl Pro to conduct meetings over zoom.
+
+## The setup instructions are below
+- Find the best spot to place the owl, ideally at the center of the table.
+- At the bottom of the device plug in the USB cable and the power adapter
+- Plug the USB cable into your laptop or computer
+- Plug the owl into an outlet. As the device starts you will hear a hoot and see the eyes of the owl illuminate.
+- Open your xoom meeting on the device used to plug in the own and change the microphone and speaker to Meeting Owl.
+- This can be done my clicking on the arrow next to the microphone icon.
+- Similarly, select the Meeting Owl as you camera by clicking on the arrow next to the video camera icon.
+- You are all set for the meeting!
diff --git a/docs/courses/NOTOS/alphafold.md b/docs/courses/NOTOS/alphafold.md
new file mode 100644
index 0000000..5ce6f43
--- /dev/null
+++ b/docs/courses/NOTOS/alphafold.md
@@ -0,0 +1,11 @@
+---
+layout: default
+title: Alphafold on Notos server
+nav_order: 5
+parent: Lab Documentation
+has_children: true
+---
+
+# **Alphafold on Notos Sever**
+
+[Luque Lab](https://www.luquelab.com/team.html) uses [Alphafold](https://github.com/deepmind/alphafold) for protein folding. AlphaFold is an artificial intelligence system developed by Google subsidiary DeepMind. It is designed to predict the three-dimensional structure of proteins, which are essential components of all living organisms. AlphaFold uses machine learning algorithms to analyze the amino acid sequences of proteins and make predictions about their shapes and structures. It represents a significant breakthrough in the field of computational biology and has the potential to greatly advance our understanding of biology and disease. This page documents the SDSU lab's endeavor to install Alphafold. The procedures outlined herein reflect the latest information as of March 30, 2023. Please note that some of the issues described here may have been resolved in subsequent updates to Alphafold, so exercise caution when using this information.
diff --git a/docs/courses/NOTOS/running_multiple_files.md b/docs/courses/NOTOS/running_multiple_files.md
new file mode 100644
index 0000000..cdabec0
--- /dev/null
+++ b/docs/courses/NOTOS/running_multiple_files.md
@@ -0,0 +1,45 @@
+---
+layout: default
+title: Running multiple protein strctures on Alphafold
+nav_order: 3
+parent: Alphafold on Notos server
+grand_parent: Lab Documentation
+has_children: false
+---
+
+# Predicting multiple protein structures using Alphafold on Notos
+
+### Setup
+1. Copy the alphafold folder from /mnt/beegfs/alphafold/alphafold to your home directory
+ * When you log in to notos you are automatically in your home directory
+ * Run the command below to copy the folder into your home directory
+ * ``` cp -r /mnt/beegfs/alphafold/alphafold/ . ```
+ * This folder has all the required updates to run alphafold successfully
+2. Locate the folder named docker by running the command ``` cd alphafold/docker ```
+3. Open run_docker.py by running the command ``` vi run_docker.py ```
+ * Press i to insert and make edits to the file
+ * Change the destination folder output_dir on line 49 to a folder you wish the results are saved into
+ * Make sure you have sufficient read and write permissions for the output_dir folder
+
+### Running multiple files on Alphafold in series on a single GPU
+1. Create a folder with all the sequences you want to run on a single GPU
+2. Check the status of the GPU's by running the command ``` nvidia-smi ```
+3. cd into your home directory using the command ``` cd ```
+4. Locate and enter the folder GPU_scripts by running the command ``` cd alphafold/GPU_scripts ```
+5. List contents in the folder by using the command ``` ls ```. The folders are named ```GPU_[GPU_number].sh ```. Choose the GPU number which isn't being used (Following step 2)
+6. Start a named screen session, by typing the following in your console ```screen -S session_name ```
+7. Run the following command to run the sequences on a single GPU
+
+ ``` bash GPU_[GPU_number].sh path_to_the_folder_with_sequences```
+
+ For example, if you want to run the sequences on GPU number 6: ``` bash GPU_6.sh ~\sequences\test_folder\``` where ``` ~\sequences\test_folder\``` is the folder with sequences.
+
+ - These scripts run alphafold using full databases. For optimized performance use the preset reduced_dbs. Open the GPU_[number] script and add the preset ``` --db_preset=reduced_dbs``` to line 9
+ - Other presets and options can be found [here](https://luquelab.github.io/Athena/courses/NOTOS/alphafold.html)
+8. Detach from the linux screen session, by press ``` Ctrl+a d ```
+ - To get the list of current running sessions, type the following ``` screen -ls ```
+ - To reattach to a linux screen, type the following ``` screen -r session_name```
+
+### Running multiple files on Alphafold in series on a multiple GPUs
+1. Create seperate folders for each set of sequences you want to run on the GPUs
+2. Repeat steps 2 to 8 for each of the folders on seperate GPUs
diff --git a/docs/courses/NOTOS/running_single_protein.md b/docs/courses/NOTOS/running_single_protein.md
new file mode 100644
index 0000000..24d900c
--- /dev/null
+++ b/docs/courses/NOTOS/running_single_protein.md
@@ -0,0 +1,42 @@
+---
+layout: default
+title: Running a single protein structure on Alphafold
+nav_order: 2
+parent: Alphafold on Notos server
+grand_parent: Lab Documentation
+has_children: false
+---
+
+# Predicting a single protein structure using Alphafold on Notos
+
+### Setup
+1. Copy the alphafold folder from /mnt/beegfs/alphafold/alphafold to your home directory
+ * When you log in to notos you are automatically in your home directory
+ * Run the command below to copy the folder into your home directory
+ * ``` cp -r /mnt/beegfs/alphafold/alphafold/ . ```
+ * This folder has all the required updates to run alphafold successfully
+2. Locate the folder named docker by running the command ``` cd alphafold/docker ```
+3. Open run_docker.py by running the command ``` vi run_docker.py ```
+ * Press i to insert and make edits to the file
+ * Change the destination folder output_dir on line 49 to a folder you wish the results are saved into
+ * Make sure you have sufficient read and write permissions for the output_dir folder
+
+### Running Alphafold
+1. To run alphafold, go back to the home directory by running the command ``` cd ```
+2. Type the command ``` python3 ~/alphafold/docker/run_docker.py --fasta_paths=path_to_sequence --max_template_date=YYYY-MM-DD --data_dir=/mnt/beegfs/alphafold/databases ```
+ * For example: ``` python3 ~/alphafold/docker/run_docker.py --fasta_paths=T1050.fasta --max_template_date=2020-05-14 --data_dir=/mnt/beegfs/alphafold/databases ```
+ * For optimized performance use the preset reduced_dbs. For example: ``` python3 ~/alphafold/docker/run_docker.py --fasta_paths=T1050.fasta --max_template_date=2020-05-14 --model_preset=monomer --db_preset=reduced_dbs --data_dir=data_dir=/mnt/beegfs/alphafold/databases ```
+ * You can use the date variable for the max_template_date by running the command ```today = `date +%Y-%m-%d` ``` and setting max_template_date to ``` max_template_date = $today ```
+3. Other presets and options can be found [here](https://luquelab.github.io/Athena/courses/NOTOS/alphafold.html)
+
+## Using a GNU screen
+To be able to detach and disconnect from a process without interrupting the procedure use GNU screen sessions:
+
+1. Follow the setup process above
+2. To start a named screen session, type the following in your console ```screen -S session_name ```
+3. Run the required alphafold commands
+4. To detach from a linux screen session, press ``` Ctrl+a d ```
+5. To get the list of current running sessions, type the following ``` screen -ls ```
+6. To reattach to a linux screen, type the following ``` screen -r session_name```
+7. More on GNU screen sessions can be found [here](https://linuxize.com/post/how-to-use-linux-screen/).
+
diff --git a/docs/courses/NOTOS/setup_alphafold.md b/docs/courses/NOTOS/setup_alphafold.md
new file mode 100644
index 0000000..7a618f1
--- /dev/null
+++ b/docs/courses/NOTOS/setup_alphafold.md
@@ -0,0 +1,564 @@
+---
+layout: default
+title: Setting up Alphafold
+nav_order: 1
+parent: Alphafold on Notos server
+grand_parent: Lab Documentation
+has_children: false
+---
+
+# AlphaFold on Notos
+
+This document provides the implementation of the pipeline of [AlphaFold V2.0](https://github.com/deepmind/alphafold) on the notos server at the Computational Science Research Center, SDSU. The notos server is managed by Dr. Christopher Paolini ([Email](paolini@engineering.sdsu.edu)).
+
+There are two CPU sockets on notos. Since, notos is not a cluster with compute nodes there is no batch scheduler. Scripts/ commands can be run on multiple GNU Screen session. More instructions are given at the end of this document. Follow [Running AlphaFold on GNU Screen session](#Running-Multiple-files-on-Alphafold).
+
+More information about AlphaFold can be found [here](https://github.com/deepmind/alphafold/blob/main/README.md).
+
+---
+## First time setup
+
+The following steps are required in order to run AlphaFold:
+
+1. Install [Docker](https://www.docker.com/).
+ * Install
+ [NVIDIA Container Toolkit](https://docs.nvidia.com/datacenter/cloud-native/container-toolkit/install-guide.html)
+ for GPU support.
+ * Setup running
+ [Docker as a non-root user](https://docs.docker.com/engine/install/linux-postinstall/#manage-docker-as-a-non-root-user).
+ * **Docker and NVIDIA Container Toolkit have been installed on notos and are up-to date as of 6/9/2022. As each new user follows the steps below to run alphafold they must contact Dr. Christopher Paolini ([Email](paolini@engineering.sdsu.edu)) for docker access.**
+1. Download genetic databases and model parameters.
+ * *The genetic databases and model parameters have been downloaded and can be found on the notos server at*
+ ```bash
+ /mnt/beegfs/alphafold/databases
+ ```
+1. Check that AlphaFold will be able to use a GPU by running:
+
+ ```bash
+ docker run --rm --gpus all nvidia/cuda:11.6.0-base-ubuntu20.04 nvidia-smi
+ ```
+
+ The output of this command should show a list of your GPUs.
+
+---
+
+## Genetic databases - Downloaded on Notos
+
+This step requires `aria2c` which is already installed and is available on the notos server.
+
+AlphaFold needs multiple genetic (sequence) databases to run:
+
+* [BFD](https://bfd.mmseqs.com/),
+* [MGnify](https://www.ebi.ac.uk/metagenomics/),
+* [PDB70](http://wwwuser.gwdg.de/~compbiol/data/hhsuite/databases/hhsuite_dbs/),
+* [PDB](https://www.rcsb.org/) (structures in the mmCIF format),
+* [PDB seqres](https://www.rcsb.org/) – only for AlphaFold-Multimer,
+* [Uniclust30](https://uniclust.mmseqs.com/),
+* [UniProt](https://www.uniprot.org/uniprot/) – only for AlphaFold-Multimer,
+* [UniRef90](https://www.uniprot.org/help/uniref).
+
+A script `scripts/download_all_data.sh` is provided on the official alphafold readme where you can download and set up all of the databases. *This script did not work as intended on the notos server. A workaround is to manually download all the databases using the scripts available at* `scripts/` *on the [Alphafold github respository](https://github.com/deepmind/alphafold)*
+
+ The `$DOWNLOAD_DIR` on notos for alphafold is `/mnt/beegfs/alphafold/databases`
+
+* Default:
+
+ ```bash
+ bash scripts/download_alphafold_params.sh
+
+ bash scripts/download_bfd.sh
+
+ bash scripts/download_mgnify.sh
+
+ bash scripts/download_pdb70.sh
+
+ bash scripts/download_pdb_mmcif.sh
+
+ bash scripts/download_pdb_seqres.sh
+
+ bash scripts/download_uniclust30.sh
+
+ bash scripts/download_uniprot.sh
+
+ bash scripts/download_uniref90.sh
+
+ ```
+
+* With `reduced_dbs`:
+
+ ```bash
+ bash scripts/download_small_bfd.sh
+ ```
+ instead of `download_bfd.sh`, will download a reduced version of the databases to be used with the
+ `reduced_dbs` database preset.
+
+**Note: The download directory `` should _not_ be a
+subdirectory in the AlphaFold repository directory.** If it is, the Docker build
+will be slow as the large databases will be copied during the image creation.
+
+**Note: The total download size for the full databases is around 415 GB
+and the total size when unzipped is 2.2 TB.**
+
+Once the script has finished, you should have the following directory structure:
+
+```
+$DOWNLOAD_DIR/ # Total: ~ 2.2 TB (download: 438 GB)
+ bfd/ # ~ 1.7 TB (download: 271.6 GB)
+ # 6 files.
+ mgnify/ # ~ 64 GB (download: 32.9 GB)
+ mgy_clusters_2018_12.fa
+ params/ # ~ 3.5 GB (download: 3.5 GB)
+ # 5 CASP14 models,
+ # 5 pTM models,
+ # 5 AlphaFold-Multimer models,
+ # LICENSE,
+ # = 16 files.
+ pdb70/ # ~ 56 GB (download: 19.5 GB)
+ # 9 files.
+ pdb_mmcif/ # ~ 206 GB (download: 46 GB)
+ mmcif_files/
+ # About 180,000 .cif files.
+ obsolete.dat
+ pdb_seqres/ # ~ 0.2 GB (download: 0.2 GB)
+ pdb_seqres.txt
+ small_bfd/ # ~ 17 GB (download: 9.6 GB)
+ bfd-first_non_consensus_sequences.fasta
+ uniclust30/ # ~ 86 GB (download: 24.9 GB)
+ uniclust30_2018_08/
+ # 13 files.
+ uniprot/ # ~ 98.3 GB (download: 49 GB)
+ uniprot.fasta
+ uniref90/ # ~ 58 GB (download: 29.7 GB)
+ uniref90.fasta
+```
+*The genetic databases and model parameters have been downloaded and can be found on the notos server at*
+
+ ```bash
+ /mnt/beegfs/alphafold/databases
+ ```
+
+---
+
+## Setting up Alphafold for the first time
+
+1. Clone this repository and `cd` into it.
+
+ ```bash
+ git clone https://github.com/deepmind/alphafold.git
+ ```
+1. Build the Docker image (Need to be done only once):
+
+ ```bash
+ docker build -f docker/Dockerfile -t alphafold .
+ ```
+
+ If you encounter the following error:
+
+ ```
+ W: GPG error: https://developer.download.nvidia.com/compute/cuda/repos/ubuntu1804/x86_64 InRelease: The following signatures couldn't be verified because the public key is not available: NO_PUBKEY A4B469963BF863CC
+ E: The repository 'https://developer.download.nvidia.com/compute/cuda/repos/ubuntu1804/x86_64 InRelease' is not signed.
+ ```
+
+ Add the following line to the `docker/Dockerfile`:
+ ```bash
+ RUN apt-key adv --fetch-keys https://developer.download.nvidia.com/compute/cuda/repos/ubuntu1804/x86_64/3bf863cc.pub
+ ```
+
+ If you encounter the following in Step 3/19:
+
+ ```
+ Step 3/9: ARG CUDA
+ symlink /proc/mounts /mnt/beegfs/notos/docker/aufs/mnt/ -inti/etc/mtab: device or resource is busy
+ ```
+ This means that all the disk space for docker is exhausted. Contact Dr. Christopher Paolini ([Email](paolini@engineering.sdsu.edu)) to add more disk space for docker.
+
+1. Install the `run_docker.py` dependencies. Note: You may optionally wish to
+ create a
+ [Python Virtual Environment](https://docs.python.org/3/tutorial/venv.html)
+ to prevent conflicts with your system's Python environment.
+
+ ```bash
+ pip3 install -r docker/requirements.txt
+ ```
+
+1. Open `docker/run_docker.py` and change the output directory `output_dir` to your choice of directory where you have sufficient permissions to write into it.
+
+1. Run `run_docker.py` pointing to a FASTA file containing the protein
+ sequence(s) for which you wish to predict the structure. If you are
+ predicting the structure of a protein that is already in PDB and you wish to
+ avoid using it as a template, then `max_template_date` must be set to be
+ before the release date of the structure. You must also provide the path to
+ the directory containing the downloaded databases. For example, for the
+ T1050 CASP14 target:
+
+ ```bash
+ python3 docker/run_docker.py \
+ --fasta_paths=T1050.fasta \
+ --max_template_date=2020-05-14 \
+ --data_dir=$DOWNLOAD_DIR
+ ```
+
+ The `$DOWNLOAD_DIR` on notos for alphafold is `/mnt/beegfs/alphafold/databases`
+
+
+ If you encounter the following error:
+
+ ```bash
+ 'Jackhmmer failed\nstderr:\n%s\n' % stderr.decode('utf-8'))
+ RuntimeError: Jackhmmer failed
+ stderr:
+ Fatal exception (source file esl_msafile_stockholm.c, line 1263):
+ stockholm msa wrote failed
+ system error: No space left on device
+ ```
+ Contact Dr. Christopher Paolini ([Email](paolini@engineering.sdsu.edu)) to allocated more space in the docker:
+
+
+ If you encounter the following error:
+
+ ```
+ TypeError: Descriptors cannot not be created directly.
+ If this call came from a _pb2.py file, your generated code is out of date and must be regenerated with protoc >= 3.19.0.
+ If you cannot immediately regenerate your protos, some other possible workarounds are:
+ 1. Downgrade the protobuf package to 3.20.x or lower.
+ 2. Set PROTOCOL_BUFFERS_PYTHON_IMPLEMENTATION=python (but this will use pure-Python parsing and will be much slower).
+ ```
+
+ Do
+ ```bash
+ pip3 install --upgrade protobuf==3.20.0
+ ```
+ And add the following line in `docker/run_docker.py` in the environmnet definition as shown below:
+
+ ```python
+ 'PROTOCOL_BUFFERS_PYTHON_IMPLEMENTATION': 'python',
+ ```
+
+ ```python
+ container = client.containers.run(
+ image=FLAGS.docker_image_name,
+ command=command_args,
+ device_requests=device_requests,
+ remove=True,
+ detach=True,
+ mounts=mounts,
+ user=FLAGS.docker_user,
+ environment={
+ 'NVIDIA_VISIBLE_DEVICES': FLAGS.gpu_devices,
+ # The following flags allow us to make predictions on proteins that
+ # would typically be too long to fit into GPU memory.
+ 'TF_FORCE_UNIFIED_MEMORY': '1',
+ 'XLA_PYTHON_CLIENT_MEM_FRACTION': '4.0',
+ 'PROTOCOL_BUFFERS_PYTHON_IMPLEMENTATION': 'python',
+ })
+ ```
+
+ If you encounter the following error:
+ ```bash
+ RuntimeError: HHblits failed
+ stdout:
+
+ stderr:
+
+ 04:13:23.681 ERROR: Could find neither hhm_db nor a3m_db!
+ ```
+ It is due to the change in permissions. Do,
+
+ ```bash
+ sudo find /mnt/beegfs/alphafold/databases -type d -exec chmod 755 {} \;
+ sudo find /mnt/beegfs/alphafold/databases -type f -exec chmod 644 {} \;
+ ```
+ If you don't have access to change permissions contact Dr. Christopher Paolini ([Email](paolini@engineering.sdsu.edu)).
+
+---
+
+## Running AlphaFold
+1. You can control which AlphaFold model to run by adding the
+ `--model_preset=` flag.
+
+ * **monomer**: This is the original model used at CASP14 with no ensembling.
+
+ * **monomer\_casp14**: This is the original model used at CASP14 with
+ `num_ensemble=8`, matching our CASP14 configuration. This is largely
+ provided for reproducibility as it is 8x more computationally
+ expensive for limited accuracy gain (+0.1 average GDT gain on CASP14
+ domains).
+
+ * **monomer\_ptm**: This is the original CASP14 model fine tuned with the
+ pTM head, providing a pairwise confidence measure. It is slightly less
+ accurate than the normal monomer model.
+
+ * **multimer**: This is the [AlphaFold-Multimer](#citing-this-work) model.
+ To use this model, provide a multi-sequence FASTA file. In addition, the
+ UniProt database should have been downloaded.
+
+1. You can control MSA speed/quality tradeoff by adding
+ `--db_preset=reduced_dbs` or `--db_preset=full_dbs` to the run command. The following presets are provided.
+
+ * **reduced\_dbs**: This preset is optimized for speed and lower hardware
+ requirements. It runs with a reduced version of the BFD database.
+ It requires 8 CPU cores (vCPUs), 8 GB of RAM, and 600 GB of disk space.
+
+ * **full\_dbs**: This runs with all genetic databases used at CASP14.
+
+ Running the command above with the `monomer` model preset and the
+ `reduced_dbs` data preset would look like this:
+
+ ```bash
+ python3 docker/run_docker.py \
+ --fasta_paths=T1050.fasta \
+ --max_template_date=2020-05-14 \
+ --model_preset=monomer \
+ --db_preset=reduced_dbs \
+ --data_dir=$DOWNLOAD_DIR
+ ```
+ The `$DOWNLOAD_DIR` on notos for alphafold is `/mnt/beegfs/alphafold/databases`
+
+---
+
+## Running AlphaFold-Multimer
+
+1. All steps are the same as when running the monomer system, but you will have to
+
+ * provide an input fasta with multiple sequences,
+ * set `--model_preset=multimer`,
+
+ An example that folds a protein complex `multimer.fasta`:
+
+ ```bash
+ python3 docker/run_docker.py \
+ --fasta_paths=multimer.fasta \
+ --max_template_date=2020-05-14 \
+ --model_preset=multimer \
+ --data_dir=$DOWNLOAD_DIR
+ ```
+ The `$DOWNLOAD_DIR` on notos for alphafold is `/mnt/beegfs/alphafold/databases`
+
+By default the multimer system will run 5 seeds per model (25 total predictions)
+for a small drop in accuracy you may wish to run a single seed per model. This
+can be done via the `--num_multimer_predictions_per_model` flag, e.g. set it to
+`--num_multimer_predictions_per_model=1` to run a single seed per model.
+
+---
+
+## Examples
+
+Below are examples on how to use AlphaFold in different scenarios.
+
+#### Folding a monomer
+
+Say we have a monomer with the sequence ``. The input fasta should be:
+
+```fasta
+>sequence_name
+
+```
+
+Then run the following command:
+
+```bash
+python3 docker/run_docker.py \
+ --fasta_paths=monomer.fasta \
+ --max_template_date=2021-11-01 \
+ --model_preset=monomer \
+ --data_dir=$DOWNLOAD_DIR
+```
+The `$DOWNLOAD_DIR` on notos for alphafold is `/mnt/beegfs/alphafold/databases`
+
+#### Folding a homomer
+
+Say we have a homomer with 3 copies of the same sequence
+``. The input fasta should be:
+
+```fasta
+>sequence_1
+
+>sequence_2
+
+>sequence_3
+
+```
+
+Then run the following command:
+
+```bash
+python3 docker/run_docker.py \
+ --fasta_paths=homomer.fasta \
+ --max_template_date=2021-11-01 \
+ --model_preset=multimer \
+ --data_dir=$DOWNLOAD_DIR
+```
+The `$DOWNLOAD_DIR` on notos for alphafold is `/mnt/beegfs/alphafold/databases`
+
+#### Folding a heteromer
+
+Say we have an A2B3 heteromer, i.e. with 2 copies of
+`` and 3 copies of ``. The input fasta should be:
+
+```fasta
+>sequence_1
+
+>sequence_2
+
+>sequence_3
+
+>sequence_4
+
+>sequence_5
+
+```
+
+Then run the following command:
+
+```bash
+python3 docker/run_docker.py \
+ --fasta_paths=heteromer.fasta \
+ --max_template_date=2021-11-01 \
+ --model_preset=multimer \
+ --data_dir=$DOWNLOAD_DIR
+```
+The `$DOWNLOAD_DIR` on notos for alphafold is `/mnt/beegfs/alphafold/databases`
+
+#### Folding multiple monomers one after another
+
+Say we have a two monomers, `monomer1.fasta` and `monomer2.fasta`.
+
+Both can be folded sequentially by using the following command:
+
+```bash
+python3 docker/run_docker.py \
+ --fasta_paths=monomer1.fasta,monomer2.fasta \
+ --max_template_date=2021-11-01 \
+ --model_preset=monomer \
+ --data_dir=$DOWNLOAD_DIR
+```
+The `$DOWNLOAD_DIR` on notos for alphafold is `/mnt/beegfs/alphafold/databases`
+
+#### Folding multiple multimers one after another
+
+Say we have a two multimers, `multimer1.fasta` and `multimer2.fasta`.
+
+Both can be folded sequentially by using the following command:
+
+```bash
+python3 docker/run_docker.py \
+ --fasta_paths=multimer1.fasta,multimer2.fasta \
+ --max_template_date=2021-11-01 \
+ --model_preset=multimer \
+ --data_dir=$DOWNLOAD_DIR
+```
+The `$DOWNLOAD_DIR` on notos for alphafold is `/mnt/beegfs/alphafold/databases`
+
+---
+
+## AlphaFold output
+
+The outputs will be saved in a subdirectory of the directory provided via the
+`--output_dir` flag of `run_docker.py` (defaults to `/tmp/alphafold/`). The
+outputs include the computed MSAs, unrelaxed structures, relaxed structures,
+ranked structures, raw model outputs, prediction metadata, and section timings.
+The `--output_dir` directory will have the following structure:
+
+```
+/
+ features.pkl
+ ranked_{0,1,2,3,4}.pdb
+ ranking_debug.json
+ relaxed_model_{1,2,3,4,5}.pdb
+ result_model_{1,2,3,4,5}.pkl
+ timings.json
+ unrelaxed_model_{1,2,3,4,5}.pdb
+ msas/
+ bfd_uniclust_hits.a3m
+ mgnify_hits.sto
+ uniref90_hits.sto
+```
+
+The contents of each output file are as follows:
+
+* `features.pkl` – A `pickle` file containing the input feature NumPy arrays
+ used by the models to produce the structures.
+* `unrelaxed_model_*.pdb` – A PDB format text file containing the predicted
+ structure, exactly as outputted by the model.
+* `relaxed_model_*.pdb` – A PDB format text file containing the predicted
+ structure, after performing an Amber relaxation procedure on the unrelaxed
+ structure prediction (see Jumper et al. 2021, Suppl. Methods 1.8.6 for
+ details).
+* `ranked_*.pdb` – A PDB format text file containing the relaxed predicted
+ structures, after reordering by model confidence. Here `ranked_0.pdb` should
+ contain the prediction with the highest confidence, and `ranked_4.pdb` the
+ prediction with the lowest confidence. To rank model confidence, we use
+ predicted LDDT (pLDDT) scores (see Jumper et al. 2021, Suppl. Methods 1.9.6
+ for details).
+* `ranking_debug.json` – A JSON format text file containing the pLDDT values
+ used to perform the model ranking, and a mapping back to the original model
+ names.
+* `timings.json` – A JSON format text file containing the times taken to run
+ each section of the AlphaFold pipeline.
+* `msas/` - A directory containing the files describing the various genetic
+ tool hits that were used to construct the input MSA.
+* `result_model_*.pkl` – A `pickle` file containing a nested dictionary of the
+ various NumPy arrays directly produced by the model. In addition to the
+ output of the structure module, this includes auxiliary outputs such as:
+
+ * Distograms (`distogram/logits` contains a NumPy array of shape [N_res,
+ N_res, N_bins] and `distogram/bin_edges` contains the definition of the
+ bins).
+ * Per-residue pLDDT scores (`plddt` contains a NumPy array of shape
+ [N_res] with the range of possible values from `0` to `100`, where `100`
+ means most confident). This can serve to identify sequence regions
+ predicted with high confidence or as an overall per-target confidence
+ score when averaged across residues.
+ * Present only if using pTM models: predicted TM-score (`ptm` field
+ contains a scalar). As a predictor of a global superposition metric,
+ this score is designed to also assess whether the model is confident in
+ the overall domain packing.
+ * Present only if using pTM models: predicted pairwise aligned errors
+ (`predicted_aligned_error` contains a NumPy array of shape [N_res,
+ N_res] with the range of possible values from `0` to
+ `max_predicted_aligned_error`, where `0` means most confident). This can
+ serve for a visualisation of domain packing confidence within the
+ structure.
+
+The pLDDT confidence measure is stored in the B-factor field of the output PDB
+files (although unlike a B-factor, higher pLDDT is better, so care must be taken
+when using for tasks such as molecular replacement).
+
+---
+## Running Multiple files on Alphafold
+
+1. To start a screen session, type `screen` in your console:
+
+```bash
+$ screen
+```
+
+1. To get a list of commands `Ctrl+a ?`
+
+1. To create a named session
+```bash
+$ screen -S session_name
+```
+
+Named sessions are useful when you run multiple screen sessions.
+
+3. To detach from a Linux screen session, press `Ctrl+a d`
+
+4. To reattach to a linux screen
+```bash
+$ screen -r
+```
+5. To get the list of current running session
+```bash
+$ screen -ls
+```
+
+More on GNU screen sessions can be found [here](https://linuxize.com/post/how-to-use-linux-screen/)
+
+---
+
+## References
+- Jumper, John, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Kathryn Tunyasuvunakool, κ.ά. ‘Highly accurate protein structure prediction with AlphaFold’. Nature 596, τχ. 7873 (2021): 583–89. https://doi.org/10.1038/s41586-021-03819-2.
+
+- Evans, Richard, Michael O\textquoterightNeill, Alexander Pritzel, Natasha Antropova, Andrew Senior, Tim Green, Augustin Žídek, κ.ά. ‘Protein complex prediction with AlphaFold-Multimer’. bioRxiv, 2021. https://doi.org/10.1101/2021.10.04.463034.
+
diff --git a/docs/courses/SDSURF.md b/docs/courses/SDSURF.md
new file mode 100644
index 0000000..1521cc5
--- /dev/null
+++ b/docs/courses/SDSURF.md
@@ -0,0 +1,15 @@
+---
+layout: default
+title: SDSURF
+parent: Lab Documentation
+nav_order: 7
+---
+
+# San Diego State University Research Foundation (SDSURF)
+
+This page summarizes the common tasks that the lab needs to develop when using research funds administered by the [SDSURF](https://foundation.sdsu.edu).
+
+## Disbursement request
++ Fill the excell spreadsheet [form](./purchasing_purch_req_standard.xlsx), including the Fund number and Account Code (for example, 7050 for supplies).
++ Upload the form and receipts to [MyRF Document Processing system](https://myrf.sdsu.edu/prod/f?p=705:LOGIN::::::).
++ Expenses for more than $1K or computer items are discouraged. A purchase requisition should be used for these items.
diff --git a/docs/courses/Work_orders.md b/docs/courses/Work_orders.md
new file mode 100644
index 0000000..c9d9507
--- /dev/null
+++ b/docs/courses/Work_orders.md
@@ -0,0 +1,15 @@
+---
+layout: default
+title: Work Orders for Luque Lab
+nav_order: 11
+parent: Lab Documentation
+---
+
+# Placing a work order at the Luque Lab
+
+- Submit a formal work order via [Facilities Services](https://bfa.sdsu.edu/campus/facilities/services).
+- Select [Submit Work Request SDSUid Required](https://sdsu.assetworks.cloud/)
+- Once logged in, choose the service needed. Fill in all the necessary details regarding the contact information, building location, and details about the issue.
+- After submitting the request online, call work control (4-4754) to report the issue. Mention the room number and that it is the Biomath lab (Luque, George, and Vaidya as PIs).
+
+- **If it is an emergency, inform [Emily Olmsted](erolmsted@sdsu.edu) in the department and keep her in the loop.**
diff --git a/docs/courses/capsid_workshop_fall_2021/capsid_anatomy_session_2021-10-26.md b/docs/courses/capsid_workshop_fall_2021/capsid_anatomy_session_2021-10-26.md
new file mode 100644
index 0000000..792a9cc
--- /dev/null
+++ b/docs/courses/capsid_workshop_fall_2021/capsid_anatomy_session_2021-10-26.md
@@ -0,0 +1,44 @@
+---
+layout: default
+title: Session 1
+nav_order: 1
+parent: Capsid Anatomy Workshop 2021
+grand_parent: Lab Documentation
+published: true
+---
+
+# Session 1: Capsid molecular model from cryoEM map (White lab)
+
+## Recorded zoom session
++ [Public recording with integrated audio transcript.](https://SDSU.zoom.us/rec/share/w3gXBfrrL0ut-_Gv1pfw0nupA8_EhQjHWFEcq4aoGnXtCfnfRKxB-lFukMhkmChU.cuKmYYbXrLwjBkTk)
+
+## Agenda
+
+### Part 1: Model building
+
+Brief overview of maps:
+
+Mention differences between cryoEM and crystallography (one is where the map is the data and the other you need to solve the structure). I would only focus on cryoEM maps. Mention it is probably worth using Isolde to re-refine existing cryoEM structures. Probably not worth it for structures worse than 4 Angstrom.
+
+Talk about where to get raw data from if you want to re-do the whole analysis (might be worthwhile for older data sets). Talk about EMDB and EMPIAR. Won’t discuss data analysis here but happy to have one-on-one sessions to teach people.
+
+Talk about phenix and CCPEM and the sharpening of cryoEM maps. CCPEM is a pain to install on mac. Also can’t handle the very large maps.
+
+Map preparation for model building:
++ Brief overview of model building
++ Show chimera and chimerax.
++ Show coot. Show how to get Oli Clarke trimmings.
++ Run through model building on coot. Talk about asymmetric unit
+
+Talk about using alphafold/ITasser/Phyre2 to make an initial model or de novo.
+
+
+### Parts 2 and 3: Model refinement
+
++ Use coot to start refine
++ Use phenix for refinement
++ Use Isolde for refinement
++ Use PDB validation server for final checking: Need to show an image of amino acid with C alpha and C beta.
+
+
+### Part 4: Overtime/questions/discussion/prolate
diff --git a/docs/courses/capsid_workshop_fall_2021/capsid_anatomy_session_2021-10-27.md b/docs/courses/capsid_workshop_fall_2021/capsid_anatomy_session_2021-10-27.md
new file mode 100644
index 0000000..9db310c
--- /dev/null
+++ b/docs/courses/capsid_workshop_fall_2021/capsid_anatomy_session_2021-10-27.md
@@ -0,0 +1,77 @@
+---
+layout: default
+title: Session 2
+nav_order: 2
+parent: Capsid Anatomy Workshop 2021
+grand_parent: Lab Documentation
+published: true
+---
+
+# Capsid Geometrical Anatomy (Luque lab)
+October 27, 2021
+
+### Zoom session
++ [Public recording with integrated audio transcript.](https://sdsu.zoom.us/rec/share/6SKUAbu6C3IYjXK-FQI5L45YlJ_xt5rO1y2ewvk4E--gEtv-_BQL5mqcaV0mgYJs.ZWoPRpkz3_gLsCBO)
+
+### Access to workshop files
++ Folder in the [Athena GitHub repo](https://github.com/luquelab/Athena/tree/gh-pages/docs/courses/capsid_workshop_fall_2021).
++ Online content provided by GitHub pages is based on the standard lab's [public documentation system](https://luquelab.github.io/Athena/courses/lab_public_documentation.html).
++ Our approach to share knowledge steams from our [Athena initiative](https://luquelab.github.io/Athena/knowledge/synthesis.html).
+
+### Temporary Athena site for the Capsid Anatomy project:
++ GitHub research repo:
++ Accessible online site:
+
+## Part 1: Capsid anatomy
+
+### Capsid molecular composition
++ Stoichiometry: Major capsid, minor capsid, scaffold, reinforcemen, and decoration proteins.
++ Functions other than major capsid protein are usually fuzzy!
+
+### Capsid physical properties
++ Diameter, surface, volume, and sphericity (internal and external).
+ + Chimera: [Icosahedron surface](https://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/icosahedron/icosahedron.html).
++ Elasticity and brittleness.
+ + No standard approach.
+
+### Capsid geometric architecture
++ T-number and icosahedral lattice; Q-number for elongated structures.
++ Generalized T-numbers: [Twarock and Luque, Nature Communications, 2019](https://doi.org/10.1038/s41467-019-12367-3).
++ Chimera X: [hkcage command](https://www.cgl.ucsf.edu/chimerax/docs/user/commands/hkcage.html) and [hkcage bundle](https://cxtoolshed.rbvi.ucsf.edu/apps/chimeraxhkcage).
++ First used in [Luque *et al.* Microorganisms, 2020](https://doi.org/10.3390/microorganisms8121944).
+
+### How to assign T-numbers
+
+**Molecular approach**: Correspondence between geometric tiles and protein clusters.
++ Example: Bacteriophage P22 (PDB 5uu5)
+ + 
+ + 
+ + 
++ Observation: *Challenge assigning minor capsid protein function:*
+
+ + 
+
+ + 
+
++ Observation: *The HK97-fold sliding hypothesis:*
+
+ + 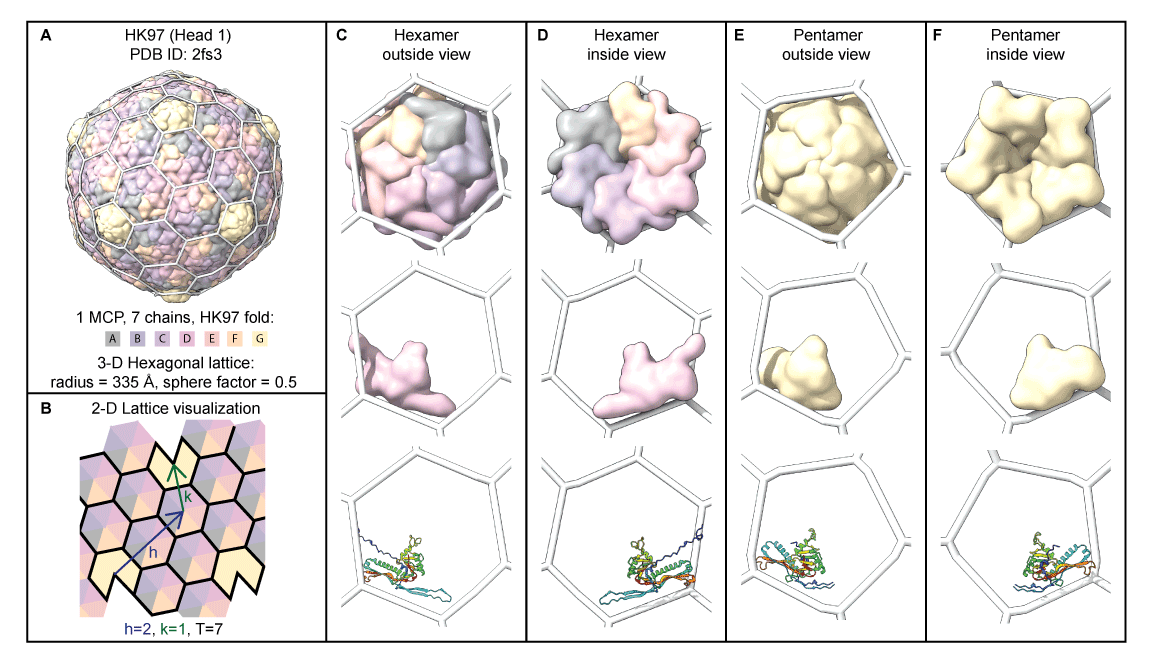
+
+ + 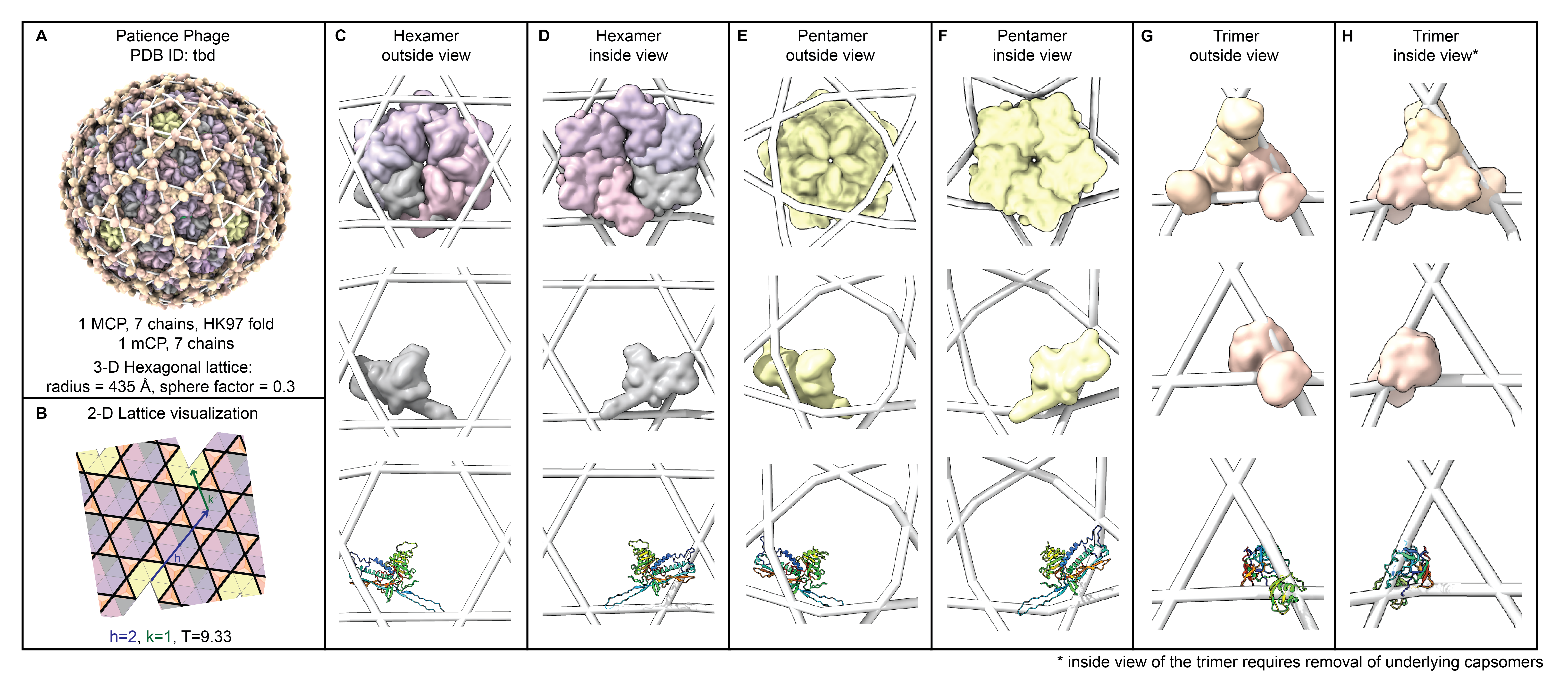
+
+ + More examples in .
+
+
+**Mechanical approach**: Correspondence between quasi-rigid regions and geometrical tiles.
++ Elastic analysis of capsids
+ + Perturbation analysis of alpha-carbon capsid models.
+ + Example: Small tobacco necrosis virus (PDB 1A34):
+ + 
+ + 
+ + Computational challenge: *large* capsids.
+
+### Part 2: Molecular approach (Diana Lee)
+
+### Part 3: Mechanical approach (Colin Brown)
+
+
diff --git a/docs/courses/capsid_workshop_fall_2021/capsid_anatomy_session_2021-11-09.md b/docs/courses/capsid_workshop_fall_2021/capsid_anatomy_session_2021-11-09.md
new file mode 100644
index 0000000..1efc0f4
--- /dev/null
+++ b/docs/courses/capsid_workshop_fall_2021/capsid_anatomy_session_2021-11-09.md
@@ -0,0 +1,16 @@
+---
+layout: default
+title: Session 3
+nav_order: 3
+parent: Capsid Anatomy Workshop 2021
+grand_parent: Lab Documentation
+published: true
+---
+
+# Capsid Geometrical Anatomy (Luque lab)
+November 9, 2021
+
+### Zoom session
++ [Zoom recording](https://sdsu.zoom.us/rec/share/OatkvkvPI51H9ehZlLRXS7d4TY4V-BMe2JpD9JmvbVBsAHuFMJF-tKDuWdxZpq9g.iQy5UXCbG9dmCJCw).
++ Agenda:
+ + Simon White led a discussion about the different groups of major capsid protein structures in the actinobacteriophage database.
diff --git a/docs/courses/capsid_workshop_fall_2021/capsid_anatomy_session_2021-11-11.md b/docs/courses/capsid_workshop_fall_2021/capsid_anatomy_session_2021-11-11.md
new file mode 100644
index 0000000..7849e67
--- /dev/null
+++ b/docs/courses/capsid_workshop_fall_2021/capsid_anatomy_session_2021-11-11.md
@@ -0,0 +1,47 @@
+---
+layout: default
+title: Session 4
+nav_order: 4
+parent: Capsid Anatomy Workshop 2021
+grand_parent: Lab Documentation
+published: true
+---
+
+# Capsid Geometrical Anatomy (Luque lab)
+November 11, 2021
+
+### Zoom session
++ [Zoom recording of Parts 2 and 3](https://sdsu.zoom.us/rec/share/F9Innzg9hiAxQi5bJz_4iTodchJRLVbtioGtWXa7g1IAoLEJnX0I7PHyHTL_s8-7.M7-yGsO2jfjRuB4F).
++ Agenda:
+ + Part 1: Toni Luque provides an update and vision about the tools that the Luque lab has been developing to predict the structural phenotype of viruses from genomic information.
+ + Part 2: Diana Lee leads a demonstration on how to predict the capsid architecture of tailed phages from their genome.
+ + Part 3: Diana Lee leads a demonstration on how to predict the capsid architecture of tailed phages from their major capsid protein gene.
+
+### Access to the main files for the session
++ [Lee et al. pre-print 2021-10-01](https://luquelab.github.io/Athena/courses/capsid_workshop_fall_2021/images/2021-11-11/Lee_et_al_manuscript_submitted_2021-10-01.pdf): This pre-print introduces the genome-to-T number and MCP-to-T number models and investigates metagenomically assembled gut phage genomes.
++ [VAN: Virus Anatomy Navigator](https://github.com/luquelab/VAN): This GitHub repository is the umbrella of the different genotype-to-phenotype viral structural tools that the Luque lab has or will develop. Checkout the **workshop** branch.
+
+## Part 1
+
+Overview. Images extracted from [Lee et al. pre-print 2021-10-01](https://luquelab.github.io/Athena/courses/capsid_workshop_fall_2021/images/2021-11-11/Lee_et_al_manuscript_submitted_2021-10-01.pdf).
+
+### Tailed phage icosahedral capsids
+
+
+### Genotype-to-phenotype approach
+
+
+### Fishing structures in metagenomes
+
+
+## Part 2
+
+### Diana Lee: Genome-to-T-number (G2T) model demonstration
+
+## Part 3
+
+### Diana Lee: MCP-to-T-number (MCP2T) model demonstration
+
+## Closure
++ Decide days for UConn visit.
++ Heads-up: Assembly Virus Workshop at SDSU (Dec 6 and 8 from 3 pm to 5 pm Pacific Time).
diff --git a/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/STNV_elastic_decomposition_giphy.gif b/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/STNV_elastic_decomposition_giphy.gif
new file mode 100644
index 0000000..978afc2
Binary files /dev/null and b/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/STNV_elastic_decomposition_giphy.gif differ
diff --git a/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/patience.png b/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/patience.png
new file mode 100644
index 0000000..593df48
Binary files /dev/null and b/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/patience.png differ
diff --git a/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/patience_inside.png b/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/patience_inside.png
new file mode 100644
index 0000000..2e64c6a
Binary files /dev/null and b/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/patience_inside.png differ
diff --git a/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/pdb_5uu5_hexagonal_lattice.png b/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/pdb_5uu5_hexagonal_lattice.png
new file mode 100644
index 0000000..b5e2269
Binary files /dev/null and b/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/pdb_5uu5_hexagonal_lattice.png differ
diff --git a/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/pdb_5uu5_trihex-dual_lattice.png b/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/pdb_5uu5_trihex-dual_lattice.png
new file mode 100644
index 0000000..1a4f112
Binary files /dev/null and b/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/pdb_5uu5_trihex-dual_lattice.png differ
diff --git a/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/pdb_5uu5_trihex_lattice.png b/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/pdb_5uu5_trihex_lattice.png
new file mode 100644
index 0000000..621e072
Binary files /dev/null and b/docs/courses/capsid_workshop_fall_2021/images/2021-10-27/pdb_5uu5_trihex_lattice.png differ
diff --git a/docs/courses/capsid_workshop_fall_2021/images/2021-11-11/Lee_et_al_manuscript_submitted_2021-10-01.pdf b/docs/courses/capsid_workshop_fall_2021/images/2021-11-11/Lee_et_al_manuscript_submitted_2021-10-01.pdf
new file mode 100644
index 0000000..09ec404
Binary files /dev/null and b/docs/courses/capsid_workshop_fall_2021/images/2021-11-11/Lee_et_al_manuscript_submitted_2021-10-01.pdf differ
diff --git a/docs/courses/capsid_workshop_fall_2021/images/2021-11-11/icosahedral_capsids_Tnumbers_2021-09-28_no_ai_capabilities.svg b/docs/courses/capsid_workshop_fall_2021/images/2021-11-11/icosahedral_capsids_Tnumbers_2021-09-28_no_ai_capabilities.svg
new file mode 100644
index 0000000..b3bec43
--- /dev/null
+++ b/docs/courses/capsid_workshop_fall_2021/images/2021-11-11/icosahedral_capsids_Tnumbers_2021-09-28_no_ai_capabilities.svg
@@ -0,0 +1,5647 @@
+
+
+
diff --git a/docs/courses/capsid_workshop_fall_2021/images/2021-11-11/methods_G2T_and_MCP2T_2021-09-28_no_ai_capabilities.svg b/docs/courses/capsid_workshop_fall_2021/images/2021-11-11/methods_G2T_and_MCP2T_2021-09-28_no_ai_capabilities.svg
new file mode 100644
index 0000000..2c7bad3
--- /dev/null
+++ b/docs/courses/capsid_workshop_fall_2021/images/2021-11-11/methods_G2T_and_MCP2T_2021-09-28_no_ai_capabilities.svg
@@ -0,0 +1,19269 @@
+
+
+
diff --git a/docs/courses/capsid_workshop_fall_2021/images/2021-11-11/predicted_T_from_gut_MCPs_2021-10-01_no_ai_capability.svg b/docs/courses/capsid_workshop_fall_2021/images/2021-11-11/predicted_T_from_gut_MCPs_2021-10-01_no_ai_capability.svg
new file mode 100644
index 0000000..1e4c6e4
--- /dev/null
+++ b/docs/courses/capsid_workshop_fall_2021/images/2021-11-11/predicted_T_from_gut_MCPs_2021-10-01_no_ai_capability.svg
@@ -0,0 +1,3943 @@
+
+
+
diff --git a/docs/courses/capsid_workshop_fall_2021/index.md b/docs/courses/capsid_workshop_fall_2021/index.md
new file mode 100644
index 0000000..9122b2d
--- /dev/null
+++ b/docs/courses/capsid_workshop_fall_2021/index.md
@@ -0,0 +1,12 @@
+---
+layout: default
+nav_order: 8
+Title: Capsid Anatomy Workshop 2021
+parent: Lab Documentation
+has_children: true
+published: true
+---
+
+# Capsid Anatomy Workshop 2021
+
+The inaugural workshop on capsid anatomy took place online in Fall 2021. The [White lab](https://scholar.google.co.uk/citations?user=ZxaXmJkAAAAJ&hl=en) (UConn) and the [Luque lab](https://www.luquelab.com) (SDSU) contributed two sessions.
diff --git a/docs/products/courses/core-readings.md b/docs/courses/core-readings.md
similarity index 96%
rename from docs/products/courses/core-readings.md
rename to docs/courses/core-readings.md
index efcc0b1..78deb48 100644
--- a/docs/products/courses/core-readings.md
+++ b/docs/courses/core-readings.md
@@ -1,8 +1,8 @@
---
layout: default
title: Core readings
-nav_order: 1
-parent: Courses and Tutorials
+nav_order: 6
+parent: Lab Documentation
---
# Core readings for lab members
diff --git a/docs/courses/googlecolab.md b/docs/courses/googlecolab.md
new file mode 100644
index 0000000..6ba0cc4
--- /dev/null
+++ b/docs/courses/googlecolab.md
@@ -0,0 +1,77 @@
+---
+layout: default
+title: Google Colab
+nav_order: 6
+parent: Lab Documentation
+---
+
+# Google Colab Instructions
+
+Google Colab is a free cloud-based platform that allows you to write and run code in your browser. It's a great tool for learning, experimenting, and collaborating on projects. This website provides you with some basic instructions to help you get started with Google Colab.
+
+## Step 1: Accessing Google Colab
+
+1. Open your web browser and go to the Google Colab website: [colab.research.google.com](https://colab.research.google.com/).
+2. You will be prompted to sign in with your Google account. If you don't have one, you can create a new account for free.
+3. Once signed in, you'll be redirected to the Google Colab homepage.
+
+## Step 2: Creating a New Notebook
+
+1. Click on the "New Notebook" button on the Google Colab homepage.
+2. A new notebook will be created, and you will be redirected to the notebook editor.
+
+## Step 3: Working with Notebooks
+
+1. Notebooks consist of cells where you can write and execute code.
+2. To add a new cell, click on the "+" button in the toolbar or press `Ctrl/Cmd + M B` for inserting a cell below or `Ctrl/Cmd + M A` for inserting a cell above.
+3. You can change the type of a cell to code or markdown by selecting the cell and clicking on the cell type dropdown in the toolbar.
+4. To execute a code cell, click on the play button on the left side of the cell or press `Ctrl/Cmd + Enter`. The code will be executed, and the output (if any) will be displayed below the cell.
+
+## Step 4: Using `pip install` in Google Colab
+
+1. Google Colab provides a pre-installed environment with many popular Python libraries. However, if you need to install additional packages, you can use the `pip install` command.
+2. To install a package, create a new code cell and type the following command:
+ ```
+ !pip install package_name
+ ```
+Replace `package_name` with the name of the package you want to install.
+
+3. After typing the command, execute the cell to install the package. The installation may take a few moments, and the output will be displayed below the cell.
+4. Once installed, you can import and use the package in subsequent code cells.
+
+## Step 5: Saving and Sharing Notebooks
+
+1. Google Colab automatically saves your notebooks to your Google Drive. You can access them anytime from the "Google Drive" tab on the left panel.
+2. To share a notebook with others, click on the "Share" button in the toolbar. You can specify the access permissions and get a shareable link.
+
+## Step 6: Using GPU and TPU
+
+1. Google Colab provides free access to GPUs and TPUs, which can greatly accelerate your code.
+2. To enable GPU or TPU acceleration, go to the "Runtime" menu, select "Change runtime type," and choose the desired accelerator from the "Hardware accelerator" dropdown.
+3. Note that the availability of GPUs and TPUs may vary depending on the current demand.
+
+These are the basic instructions to get started with Google Colab.
+
+## Additional Topics
+
+Here are some additional topics that you may find helpful when working with Google Colab:
+
+### Managing Code and Text Cells
+
+- You can add, delete, rearrange, and merge cells in Google Colab. To convert cells between code and text (markdown) formats, select the cell and click on the cell type dropdown in the toolbar.
+
+### Using Code Snippets and Code Completion
+
+- Google Colab provides code snippets and code completion features that can enhance productivity. Take advantage of these features to speed up your coding process.
+
+### Importing and Exporting Notebooks
+
+- In addition to saving notebooks to Google Drive, you can import existing notebooks into Google Colab. You can also export notebooks to different formats, such as `.ipynb` or `.py` files.
+
+### Collaboration and Version Control
+
+- Google Colab allows for real-time collaboration with others. Multiple users can work on the same notebook simultaneously. You can also use version control tools like GitHub to manage notebook versions.
+
+### Using External Data
+
+- If you need to work with external data in your notebooks, you can upload data files or connect to external data sources such as Google Drive, Google Sheets, or cloud storage services.
diff --git a/docs/products/courses/index.md b/docs/courses/index.md
similarity index 72%
rename from docs/products/courses/index.md
rename to docs/courses/index.md
index 176f6df..492c338 100644
--- a/docs/products/courses/index.md
+++ b/docs/courses/index.md
@@ -1,8 +1,7 @@
---
layout: default
-title: Courses and Tutorials
-nav_order: 3
-parent: Products
+title: Lab Documentation
+nav_order: 5
has_children: true
---
diff --git a/docs/courses/installable_python_package/conda.md b/docs/courses/installable_python_package/conda.md
new file mode 100644
index 0000000..87b7454
--- /dev/null
+++ b/docs/courses/installable_python_package/conda.md
@@ -0,0 +1,218 @@
+---
+layout: default
+title: Python Package Build Process - Conda
+nav_order: 2
+parent: Installable python package
+grand_parent: Lab Documentation
+has_children: False
+---
+
+# Building conda packages
+
+This tutorial describes how to build a conda package
+
+1. Install [Miniconda or Anaconda](https://docs.anaconda.com/anaconda/install/), conda-build and Git
+
+ - After installing miniconda or anaconda, use conda to install conda-build and Git.
+ - To install conda-build in your terminal window or an anaconda prompt, run:
+
+ ```
+ conda install conda-build
+ ```
+ - Git can be installed by following the steps mentioned [here](https://github.com/git-guides/install-git).
+ - Follow [Building conda packages with conda skeleton](https://docs.conda.io/projects/conda-build/en/latest/user-guide/tutorials/build-pkgs-skeleton.html). The lab's GitHub template already has the basic skeleton.
+
+2. Edit the [meta.yaml](https://docs.conda.io/projects/conda-build/en/latest/resources/define-metadata.html) file. It should look something like this:
+ {% raw %}
+ ```
+ {% set version = "1.1.0" %}
+
+ package:
+ name: imagesize
+ version: {{ version }}
+
+ source:
+ git_url: https://github.com/ilanschnell/bsdiff4.git
+ git_rev: 1.1.4
+ git_depth: 1 # (Defaults to -1/not shallow)
+
+ build:
+ noarch: python
+ number: 0
+ script: python -m pip install .
+
+ requirements:
+ host:
+ - python
+ - pip
+ run:
+ - python
+
+ test:
+ imports:
+ - imagesize
+
+ about:
+ home: https://github.com/shibukawa/imagesize_py
+ license: MIT
+ summary: 'Getting image size from png/jpeg/jpeg2000/gif file'
+ description: |
+ This module analyzes jpeg/jpeg2000/png/gif image header and
+ return image size.
+ dev_url: https://github.com/shibukawa/imagesize_py
+ doc_url: https://pypi.python.org/pypi/imagesize
+ doc_source_url: https://github.com/shibukawa/imagesize_py/blob/master/README.rst
+
+ ```
+ {% endraw %}
+ - Here is another example from the pyCapsid repository
+
+ {% raw %}
+ ```
+ #{% set data = load_setup_py_data() %}
+
+ package:
+ name: pycapsid
+ version: 0.1.9
+
+ source:
+ path: ..
+ #url: https://pypi.io/packages/source/{{ name[0] }}/{{ name }}/{{ name }}-{{ version }}.tar.gz
+ # If getting the source from GitHub, remove the line above,
+ # uncomment the line below, and modify as needed. Use releases if available:
+ #url: https://github.com/luquelab/pyCapsid/archive/refs/heads/colin_branch.zip
+ # and otherwise fall back to archive:
+ # url: https://github.com/simplejson/simplejson/archive/v{{ version }}.tar.gz
+ #sha256: 2b3a0c466fb4a1014ea131c2b8ea7c519f9278eba73d6fcb361b7bdb4fd494e9
+ # sha256 is the preferred checksum -- you can get it for a file with:
+ # `openssl sha256 `.
+ # You may need the openssl package, available on conda-forge:
+ # `conda install openssl -c conda-forge``
+
+ build:
+ script: python -m pip install . -vv
+ number: 1
+ # Uncomment the following line if the package is pure Python and the recipe is exactly the same for all platforms.
+ # It is okay if the dependencies are not built for all platforms/versions, although selectors are still not allowed.
+ # See https://conda-forge.org/docs/maintainer/knowledge_base.html#noarch-python for more details.
+ noarch: python
+ # If the installation is complex, or different between Unix and Windows, use separate bld.bat and build.sh files instead of this key.
+ # By default, the package will be built for the Python versions supported by conda-forge and for all major OSs.
+ # Add the line "skip: True # [py<35]" (for example) to limit to Python 3.5 and newer, or "skip: True # [not win]" to limit to Windows.
+ # More info about selectors can be found in the conda-build docs:
+ # https://docs.conda.io/projects/conda-build/en/latest/resources/define-metadata.html#preprocessing-selectors
+
+
+ requirements:
+ host:
+ - python <3.11
+ - pip
+ - setuptools
+ - setuptools-scm
+ run:
+ - python<3.11
+ - biotite
+ - scikit-learn
+ - numpy<1.24
+ - scipy
+ - matplotlib-base
+ - numba
+ - statsmodels
+
+ test:
+ # Some packages might need a `test/commands` key to check CLI.
+ # List all the packages/modules that `run_test.py` imports.
+ imports:
+ - pyCapsid
+ # For python packages, it is useful to run pip check. However, sometimes the
+ # metadata used by pip is out of date. Thus this section is optional if it is
+ # failing.
+ requires:
+ - pip
+
+ about:
+ home: https://github.com/luquelab/pycapsid
+ summary: A set of computational tools written in python for the analysis of viral capsids
+ # Remember to specify the license variants for BSD, Apache, GPL, and LGPL.
+ # Use the SPDX identifier, e.g: GPL-2.0-only instead of GNU General Public License version 2.0
+ # See https://spdx.org/licenses/
+ license: MIT
+ # The license_family, i.e. "BSD" if license is "BSD-3-Clause".
+ # Optional
+ license_family: MIT
+ # It is required to include a license file in the package,
+ # (even if the license doesn't require it) using the license_file entry.
+ # Please also note that some projects have multiple license files which all need to be added using a valid yaml list.
+ # See https://docs.conda.io/projects/conda-build/en/latest/resources/define-metadata.html#license-file
+ license_file: LICENSE.txt
+ # The doc_url and dev_url are optional.
+ #doc_url: https://simplejson.readthedocs.io/
+ #dev_url: https://github.com/simplejson/simplejson
+
+ extra:
+ recipe-maintainers:
+ - colintravisbrown
+ # # GitHub IDs for maintainers of the recipe.
+ # # Always check with the people listed below if they are OK becoming maintainers of the recipe. (There will be spam!)
+ # - LisaSimpson
+ # - LandoCalrissian
+ ```
+ {% endraw %}
+
+3. Write build script files build.sh and bls.bat if noarch: python is not included.
+ - ```build.sh```---Shell script for macOS and Linux.
+ - ```bld.bat```---Batch file for Windows.
+ - The 2 files ```build.sh``` and ```bld.bat``` must be in the same directory as your ```meta.yaml``` file.
+
+ **bld.bat**
+
+ - Type exactly as shown below in ```bld.bat```
+ ```
+ "%PYTHON%" setup.py install
+ if errorlevel 1 exit 1
+ ```
+
+ **build.sh**
+
+ - Type exactly as shown below in ```build.sh```
+ ```
+ $PYTHON setup.py install # Python command to install the script.
+
+ ```
+4. Building and Installing
+ - Run conda-build:
+ ```
+ conda-build click
+ ```
+ - If the package has dependencies, link the channel containing them when doing conda build.
+ ```
+ conda build -c conda-forge --output-folder .
+ ```
+ - When conda-build is finished, it displays the package filename and location.
+ - Install your newly built program on your local computer by using the ```use-local``` flag:
+ ```
+ conda install --use-local click
+ ```
+
+5. Uploading new packages to Anaconda.org
+ - Open an account on Anaconda.org.
+ - Run the command ```conda install anaconda-client```, enter your username and password.
+ - Log into your [Anaconda.org](http://anaconda.org/) account with the command:
+ ```
+ anaconda login
+ ```
+ - Upload your package to Anaconda.org
+ ```
+ anaconda upload ~/miniconda/conda-bld/linux-64/click-7.0-py37_0.tar.bz2
+ ```
+
+6. Installing the package after uploading it
+ - First, create a new virtual environment using conda and then activate it.
+
+ ```
+ conda create -n envName python=3.10 -y
+ conda activate envName
+ ```
+ ```
+ conda install -c account_name -c conda-forge package-name
+ ```
diff --git a/docs/courses/installable_python_package/index.md b/docs/courses/installable_python_package/index.md
new file mode 100644
index 0000000..291ee24
--- /dev/null
+++ b/docs/courses/installable_python_package/index.md
@@ -0,0 +1,13 @@
+---
+layout: default
+title: Installable python package
+nav_order: 3
+parent: Lab Documentation
+has_children: true
+---
+
+# **Installing python packages for lab members**
+
+
+
+This website contains comprehensive instructions and procedures to guide you through the process of creating a Python package using both Pip and Conda.
diff --git a/docs/courses/installable_python_package/pip.md b/docs/courses/installable_python_package/pip.md
new file mode 100644
index 0000000..160786f
--- /dev/null
+++ b/docs/courses/installable_python_package/pip.md
@@ -0,0 +1,156 @@
+---
+layout: default
+title: Python Package Build Process - PyPI
+nav_order: 1
+parent: Installable python package
+grand_parent: Lab Documentation
+has_children: False
+---
+
+# Python Package Build Process: PyPI
+The following tutorial gives the process to create a python package.
+
+1. Make sure you have the latest version of pip installed on your device. To update pip run the following command:
+
+ - Linux/Mac: ``` python3 -m pip install --upgrade pip```
+ - Windows: ``` py -m pip install --upgrade pip ```
+
+2. Begin with creating the following file structure locally. Make sure the package name here is unique. The structure is as follows:
+
+```
+project/
+└───src/
+│ └───package_name
+│ └─── __init__.py
+│ └─── example.py
+```
+
+- Continue to populate the files added:
+ - Package name that contains the python files should match the project name.
+ - ```__init__.py``` is required to import the directory as a package, and should be empty
+ - ```example.py``` is an example of a module within the package that could contain the logic (functions, classes, constants, etc.) of your package
+
+
+3. Update the project structure as follows
+```
+project/
+│ README.md
+| setup.py
+│ LICENCE
+└───src/
+│ └───package_name
+│ │ __init__.py
+│ │ example.py
+└───tests\
+```
+
+- ``` setup.py``` tells “frontend” build tools like pip and build what “backend” tool to use to create distribution packages for your project. ```setup.py``` contains the following content.
+
+ 1. name: A string containing the package’s name.
+ 2. version: A string containing the package’s version number.
+ 3. description: A single-line text explaining the package.
+ 4. author: A string identifying the package’s creator/author.
+ 5. long_description: A string containing a more detailed description of the package.
+ 6. maintainer: It's a string providing the current maintainer’s name, if not the author. If the maintainer is not given, the author in PKG-INFO will be utilized by the setup tools.
+ 7. url: A string providing the package’s homepage URL (usually the GitHub repository or the PyPI page).
+ 8. download_url: A string containing the URL where the package may be downloaded.
+ 9. package_data: This is a dictionary where the keys are package names and the values are lists of glob patterns.
+ 10. py_modules: A string list containing the modules that setup tools will modify.
+ 11. python_requires: This is a comma-separated string providing Python version specifiers for the package’s supported Python versions.
+ 12. install_requires: A string list containing only the dependencies necessary for the package to function effectively.
+ 13. keywords: A comma-separated string or string list providing descriptive meta-data.
+ 14. entry_points: This is a dictionary with keys corresponding to entry point names and values corresponding to the actual entry points stated in the source code.
+ 15. license: A string containing the package’s licensing information.
+ 16. keywords: A list of keywords.
+ 17. long_description: longer description of the package.
+
+Example:
+```
+from setuptools import setup
+setup(
+ name='app-name'
+ version='1.0',
+ author='Bejamin Frakline',
+ description='A brief synopsis of the project',
+ long_description='A much longer explanation of the project and helpful resources',
+ url='https://github.com/BenjaminFranline',
+ keywords='development, setup, setuptools',
+ python_requires='>=3.7, <4',
+ packages=find_packages(include=['exampleproject', 'exampleproject.*']),
+ install_requires=[
+ 'PyYAML',
+ 'pandas==0.23.3',
+ 'numpy>=1.14.5',
+ 'matplotlib>=2.2.0,,
+ 'jupyter'
+ ],
+ package_data={
+ 'sample': ['sample_data.csv'],
+ },
+ entry_points={
+ 'runners': [
+ 'sample=sample:main',
+ ]
+ }
+)
+```
+- Create README.md to give users a detailed description of the package
+- Create a License
+
+4. Generate distribution packages for the package. These are archives that are uploaded to the Python Package Index that can be installed by ```pip```
+ - Run the following command from the same directory where pyproject.toml is located:
+ - Linux/Mac: ``` python3 -m pip install --upgrade build ```
+ - Windows: ``` py -m build ```
+ - This command should output a lot of text and once completed should generate two files in the ```dist``` directory:
+5. Upload the distribution archives
+ - Register an account on [TestPyPI](https://test.pypi.org/account/register/),which is a separate instance of the package index intended for testing and experimentation.
+ - Use [twine](https://packaging.python.org/en/latest/key_projects/#twine) tp upload the distribution packages
+
+ - Linux/Mac: ```python3 -m pip install --upgrade twine```
+ - Windows: ```py -m pip install --upgrade twine```
+ - Run Twine to upload all the archives under ```dist```
+ - Linux/Mac: ```python3 -m twine upload --repository testpypi dist/*```
+ - Windows: ```py -m twine upload --repository testpypi dist/*```
+ - You will be prompted for a username and password. For the username, use ```__token__```. For the password, use the token value, including the pypi- prefix.
+ - Once uploaded, your package should be viewable on TestPyPI; for example: https://test.pypi.org/project/package_name.
+
+6. Installing the newly uploaded package
+- You can use pip to install your package and verify that it works. Create a virtual environment and install your package from TestPyPI:
+ - Linux/Mac: ``` python3 -m pip install --index-url https://test.pypi.org/simple/ --no-deps package_name```
+ - Windows: ``` py -m pip install --index-url https://test.pypi.org/simple/ --no-deps package_name```
+- Test if the package was installed correctly by importing the package. Make sure you are still in the virtual environment, then run python by running the command:
+ - Linux/Mac: ```python3```
+ - Windows: ```py```
+- Import the package
+ - ``` from package_name import func ```
+ - Call one of the functions in the package
+7. You've now packaged and distributed a Python project
+
+---
+
+- ```pyproject.toml``` can be used instead of ```setup.py```. Both these files serve the same purpose.
+ - Open ```pyproject.toml``` and enter the ```[build-system]``` tables: ``` [build-system]\ requires = ["hatchling"]\ build-backend = "hatchling.build" ```
+ - ```requires``` is a list of packages that are needed to build your package. You don’t need to install them; build frontends like pip will install them automatically in a temporary, isolated virtual environment for use during the build process
+ - ``build-backend``` is the name of the Python object that frontends will use to perform the build.
+ - Configure metadata as follows:
+
+```
+[project]
+name = "example_package_YOUR_USERNAME_HERE"
+version = "0.0.1"
+authors = [
+ { name="Example Author", email="author@example.com" },
+]
+description = "A small example package"
+readme = "README.md"
+requires-python = ">=3.7"
+classifiers = [
+ "Programming Language :: Python :: 3",
+ "License :: OSI Approved :: MIT License",
+ "Operating System :: OS Independent",
+]
+
+[project.urls]
+"Homepage" = "https://github.com/pypa/sampleproject"
+"Bug Tracker" = "https://github.com/pypa/sampleproject/issues"
+```
diff --git a/docs/courses/lab_public_documentation.md b/docs/courses/lab_public_documentation.md
new file mode 100644
index 0000000..efa6ee6
--- /dev/null
+++ b/docs/courses/lab_public_documentation.md
@@ -0,0 +1,31 @@
+---
+layout: default
+title: Public documentation
+nav_order: 1
+parent: Lab Documentation
+---
+
+# Sharing the lab's knowledge
+
+A core principle in the lab is to share the produced knowledge and technologies with the specialized and general audiences alike, independently of journals. The synthesis and description of the lab's research is a continuous task that must begin shaping at the very start of each project.
+
+The lab currently generates public documentation using [GitHub Pages](https://pages.github.com). [GitHub](http://github.com/) uses [Jekyll](http://jekyllrb.com/) to generate the static websites. To understand better how this works, check the [GitHub Pages documentation](https://docs.github.com/en/pages).
+
+
+## Publishing the documentation of a new project
+1. Create the repo using the [project_template](https://github.com/luquelab/project_template) repo as a template. Press the green "use this template" button, and make sure it's public.
+2. Select the "Main" dropdown on the left, and create a gh-pages branch. This branch will be dedicated to monitor the changes on the website. Once the changes are satisfactory, the modifications should be merged in the repo's main branch.
+3. To generate the website, go to the repo's Settings, click on the Pages panel. Use the /docs folder in the gh-pages branch as the website's source.
+4. GitHub will automatically generate the static website.
+5. Any commited changes to the gh-pages branch will be reflected on the static website. Unfortunately, these changes are not instantaneous for sites hosted on GitHub Pages. Be patient and minimize structural changes.
+6. To learn more about how to modify your documentation, explore the links in the theme associated with the [project template's repo site](http://luquelab.github.io/project_template/). Explore also the other public documentations from the lab. You can easily access the associated GitHub repos to mimick features that can help you in your documentation.
+
+## Publishing the documentation of an ongoing project
+1. Import the [/docs](https://github.com/luquelab/project_template/tree/main/docs) folder from the [project_template](https://github.com/luquelab/project_template) repo and adapt its structure as needed for your documentation. The Home and Synthesis sections must be kept and updated accordingly to the project.
+2. The rest of the steps are analogous to the [new project section](#publishing-the-documentation-of-a-new-project), skipping step 1.
+
+## Understanding useful advanced features
++ Customize the site modifying the [_config.yml](https://jekyllrb.com/docs/configuration/) file in /docs coded in [YAML](https://yaml.org). You will need to update the aux_links to link to your own repo, and to change "Project Template" to the name of your own project.
++ Modify the structure and dependencies of your pages adapting the [Front Matter](https://jekyllrb.com/docs/front-matter/) section in your markdown files coded also using [YAML](https://yaml.org).
++ Modify the html templates of the [theme](https://jekyllrb.com/docs/themes/) if necessary.
++ Write math using [MathJax](http://mathjax.org/), which is based on [LaTex synthax](https://en.wikibooks.org/wiki/LaTeX/Mathematics). The [project_template](https://github.com/luquelab/project_template) repo contains a modified [head.html](https://github.com/luquelab/project_template/blob/main/docs/_includes/head.html) template supporting MathJax in the website.
diff --git a/docs/courses/markdown_tutorials/add_dropdowns.md b/docs/courses/markdown_tutorials/add_dropdowns.md
new file mode 100644
index 0000000..5ea8ac8
--- /dev/null
+++ b/docs/courses/markdown_tutorials/add_dropdowns.md
@@ -0,0 +1,43 @@
+---
+layout: default
+title: Adding Dropdown Menus for GitHub pages
+nav_order: 5
+parent: GitHub for Luque Lab
+grand_parent: Lab Documentation
+---
+
+# Adding Dropdown Menus to Projects
+Dropdown menus are a helpful way of organizing information. Here, on the Athena Website, we have several dropdown menus including "Courses and Tutorials" which will serve as the example for this tutorial.
+
+On Github, we have stored a "Courses and Tutorials" folder that contains the index mark down file (index.md) and the markdown files for each of the child pages.
+
+You will first need to add the following has_children attribute to your parent page's index file. For example, on this website, "Courses and Tutorials" is the parent page, and it extends the children "Core Readings", "Github", etc., but don't worry about the children quite yet.
+
+{:width="350px"}
+
+Now that we've given the Courses and Tutorials index the has_children attribute and set it to true, a downwards pointing arrow is displayed beside it. However, when you click on the carrot, there's nothing there.
+
+{:width="200px"}
+
+We still need to add the parent attributes to the children. Open a markdown file you wish to be contained within the dropdown menu, and add the parent attribute. Check the title attribute of the parent page (in this case, the Courses and Tutorials index.md file) to make sure you are attaching the correct page.
+
+{:width="350px"}
+
+## Nested Dropdown Menus
+
+You can also create dropdown menus within dropdown menus by adding a grand_parent attribute to the grand child, and adding a has_children attribute to the child.
+
+For this example, Courses and Tutorials is the grandparent, Markdown Tutorials is the child, and Adding Dropdown Menus will be the grandchild. We will be adding the has_children attribute to the Markdown Tutorials index.md, and the grand_parent attribute to the Adding Dropdown Menus file.
+
+Here, I've added the grand_parent attribute to this page we're on (Adding Dropdown Menus). Since the initial dropdown menu was Courses and Tutorials, we will enter that as the title for the grand_parent attribute.
+
+{:width="350px"}
+
+Then I added the has_children attribute to the Markdown Tutorials index file, which is the parent of the grandchild.
+
+{:width="350px"}
+
+Now, your dropdown menus should be nested like so:
+
+{:width="250px"}
+
diff --git a/docs/courses/markdown_tutorials/add_images.md b/docs/courses/markdown_tutorials/add_images.md
new file mode 100644
index 0000000..bc3cc37
--- /dev/null
+++ b/docs/courses/markdown_tutorials/add_images.md
@@ -0,0 +1,53 @@
+---
+layout: default
+title: Adding Images to Markdown Files
+nav_order: 6
+parent: GitHub for Luque Lab
+grand_parent: Lab Documentation
+---
+
+# Adding Images to Markdown Files
+
+## SVG Files
+SVG files work best for our purposes, but JPGs and PNGs work as well. When you do use SVG files, make sure that you do not check the box to preserve illustrator editing capabilites.
+
+The process of adding images to markdown files is fairly simple. But before you start adding images to your markdown file, you need to create an images folder.
+
+To do that, navigate to the same directory your markdown file is located in, and click on "Add file", and create a new file.
+
+{:width="300px"}
+
+Name your file "images" and add a forward slash (/) at the end so that github understands you're adding a folder.
+
+{:width="500px"}
+
+Add a README.md to your images folder breifly stating where the images stored in the folder are going to be implemented.
+
+{:width="500px"}
+
+Commit the changes and navigate back to the images folder you just created.
+
+Chances are you are going to have multiple markdown pages in the same directory that will all be implementing pngs. Inside the images folder we will create a different folder for each markdown file that will be using PNGs.
+
+Name the new folder after the markdown file, and add the README.md with a brief statement about where the images will be implemented.
+
+I have named this markdown file I am currently using as "add_images" so I will name my images folder for this page "add_images".
+
+{:width="500px"}
+
+Make sure you have named your PNGs descriptively, and then drag and drop them into your folder. Commit the changes.
+
+You will use the following syntax to add your image into the markdown file:
+
+{:width="400px"}
+
+"alt_text" is what will show up on the webpage should the image fail. The file path provides the location of the PNG.
+
+If you want to include an image of a puppy in a folder named animals, your alt text might be "puppy" and your file path would be /images/animals/puppy.png
+
+{:width="400px"}
+
+However, you may want to resize the image you are using. In that case, you would use the following syntax, and would have to play around with the pixels to find a size that works for you. The image height will adjust as you adjust the image width.
+
+{:width="400px"}
+
diff --git a/docs/courses/markdown_tutorials/images/README.md b/docs/courses/markdown_tutorials/images/README.md
new file mode 100644
index 0000000..cde4356
--- /dev/null
+++ b/docs/courses/markdown_tutorials/images/README.md
@@ -0,0 +1,3 @@
+The images for the markdown tutorials are stored here.
+
+Each folder within the images folder contains the images for their respective markdown pages.
diff --git a/docs/courses/markdown_tutorials/images/add_dropdowns/README.md b/docs/courses/markdown_tutorials/images/add_dropdowns/README.md
new file mode 100644
index 0000000..a2cc714
--- /dev/null
+++ b/docs/courses/markdown_tutorials/images/add_dropdowns/README.md
@@ -0,0 +1 @@
+The images for the Add Dropdowns mark down tutorial are stored here.
diff --git a/docs/courses/markdown_tutorials/images/add_dropdowns/add_images.png b/docs/courses/markdown_tutorials/images/add_dropdowns/add_images.png
new file mode 100644
index 0000000..c980390
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_dropdowns/add_images.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_dropdowns/child_has_children.png b/docs/courses/markdown_tutorials/images/add_dropdowns/child_has_children.png
new file mode 100644
index 0000000..a902f82
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_dropdowns/child_has_children.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_dropdowns/courses.png b/docs/courses/markdown_tutorials/images/add_dropdowns/courses.png
new file mode 100644
index 0000000..38cdb43
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_dropdowns/courses.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_dropdowns/grand_parent.png b/docs/courses/markdown_tutorials/images/add_dropdowns/grand_parent.png
new file mode 100644
index 0000000..cb0b8b9
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_dropdowns/grand_parent.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_dropdowns/has_children.png b/docs/courses/markdown_tutorials/images/add_dropdowns/has_children.png
new file mode 100644
index 0000000..f63f2af
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_dropdowns/has_children.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_dropdowns/nested.png b/docs/courses/markdown_tutorials/images/add_dropdowns/nested.png
new file mode 100644
index 0000000..7097172
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_dropdowns/nested.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_dropdowns/parent.png b/docs/courses/markdown_tutorials/images/add_dropdowns/parent.png
new file mode 100644
index 0000000..f5d7bbf
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_dropdowns/parent.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_images/README.md b/docs/courses/markdown_tutorials/images/add_images/README.md
new file mode 100644
index 0000000..cb4a852
--- /dev/null
+++ b/docs/courses/markdown_tutorials/images/add_images/README.md
@@ -0,0 +1 @@
+Folder to contain images for the add_images page within markdown_tutorials.
diff --git a/docs/courses/markdown_tutorials/images/add_images/add_images.png b/docs/courses/markdown_tutorials/images/add_images/add_images.png
new file mode 100644
index 0000000..c980390
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_images/add_images.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_images/adjust_width.png b/docs/courses/markdown_tutorials/images/add_images/adjust_width.png
new file mode 100644
index 0000000..24c8817
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_images/adjust_width.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_images/folder.png b/docs/courses/markdown_tutorials/images/add_images/folder.png
new file mode 100644
index 0000000..9f9d4a1
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_images/folder.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_images/grand_parent.png b/docs/courses/markdown_tutorials/images/add_images/grand_parent.png
new file mode 100644
index 0000000..cb0b8b9
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_images/grand_parent.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_images/image_syntax.png b/docs/courses/markdown_tutorials/images/add_images/image_syntax.png
new file mode 100644
index 0000000..054dbed
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_images/image_syntax.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_images/make_folder.png b/docs/courses/markdown_tutorials/images/add_images/make_folder.png
new file mode 100644
index 0000000..5aa8123
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_images/make_folder.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_images/make_readme.png b/docs/courses/markdown_tutorials/images/add_images/make_readme.png
new file mode 100644
index 0000000..fafa9e3
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_images/make_readme.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_images/new_file.png b/docs/courses/markdown_tutorials/images/add_images/new_file.png
new file mode 100644
index 0000000..bbb63ec
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_images/new_file.png differ
diff --git a/docs/courses/markdown_tutorials/images/add_images/puppy_example.png b/docs/courses/markdown_tutorials/images/add_images/puppy_example.png
new file mode 100644
index 0000000..3c4ecf1
Binary files /dev/null and b/docs/courses/markdown_tutorials/images/add_images/puppy_example.png differ
diff --git a/docs/courses/markdown_tutorials/index.md b/docs/courses/markdown_tutorials/index.md
new file mode 100644
index 0000000..f3e407d
--- /dev/null
+++ b/docs/courses/markdown_tutorials/index.md
@@ -0,0 +1,16 @@
+---
+layout: default
+nav_order: 4
+Title: Markdown for GitHub
+parent: GitHub for Luque Lab
+grand_parent: Lab Documentation
+---
+
+# Markdown Tutorials
+
+Here are some helpful resources for Markdown's syntax:
+
+- [Markdown Guide by Matt Cone](https://www.markdownguide.org/basic-syntax/)
+- [Github Markdown Reference by Rick Cogley](https://github.com/RickCogley/Github-Markdown-Reference#links)
+- [Commonmark Quick Reference](https://commonmark.org/help/)
+
diff --git a/docs/courses/o'reilly-access-database.md b/docs/courses/o'reilly-access-database.md
new file mode 100644
index 0000000..a1151f8
--- /dev/null
+++ b/docs/courses/o'reilly-access-database.md
@@ -0,0 +1,26 @@
+---
+layout: default
+title: O'Reilly Learning
+nav_order: 9
+parent: Lab Documentation
+---
+# O'Reilly Learning
+Through San Diego State University, all students have access to [O'Reilly](https://learning.oreilly.com/home/), a database full of free books, audio books, case studies, and other resources.
+
+You can access this database directly with the following steps:
+
+
+1. Either click on [this link](https://learning.oreilly.com/home/) to access O'Reilly directly, or navigate there yourself by:
+
+ 1. Heading to the [SDSU Library website](https://library.sdsu.edu/)
+ 2. Hover over "Find" and click on "Journals & Databases"
+ 3. Select "S"
+ 4. Select "Safari eBooks Online"
+
+2. San Diego State University is not listed in the dropdown menu, so click "Not Listed? Click here".
+3. Enter your SDSUid. This is your school email that ends in "@sdsu.edu".
+4. Click on the Profile button on the top right hand corner of the screen, and select "Playlists" from the dropdown menu.
+5. Select the red "New +" button and name the playlist "Lab Reference Materials".
+6. Search for The Pragmatic Programmer: your journey to mastery, 20th Anniversary Edition, and add it to your Lab Reference Materials playlist for easy access
+
+Now you have access to 35K+ books, 30K+ hours of video, curated learning paths, case studies, interactive tutorials, audio books, and O'Reilly conference videos.
diff --git a/docs/courses/purchasing_purch_req_standard.xlsx b/docs/courses/purchasing_purch_req_standard.xlsx
new file mode 100644
index 0000000..b9a5470
Binary files /dev/null and b/docs/courses/purchasing_purch_req_standard.xlsx differ
diff --git a/docs/index.md b/docs/index.md
index 3b4de8e..ceb9e13 100644
--- a/docs/index.md
+++ b/docs/index.md
@@ -7,8 +7,11 @@ has_children: false
## The [Luque lab](https://www.luquelab.com) investigates the physical origin, ecology, and evolution of viruses.
-The lab's mission is to decipher the role that viruses played in the origin of life and use this knowledge to help humankind explore and habit new frontiers. The lab is part of the [Viral Information Institute](https://viralization.org), the [Computational Science Research Center](http://www.csrc.sdsu.edu), and the [Department of Mathematics and Statistics](https://math.sdsu.edu) at [San Diego State University](https://www.sdsu.edu).
+[Site under construction]
+
+The lab's long-term mission is to decipher the role that viruses played in the origin of life and use this knowledge to help humankind explore and habit new frontiers. The lab is part of the [Viral Information Institute](https://viralization.org), the [Computational Science Research Center](http://www.csrc.sdsu.edu), and the [Department of Mathematics and Statistics](https://math.sdsu.edu) at [San Diego State University](https://www.sdsu.edu).
The current lab approach is based on computational modeling, combining mathematics, statistics, and biophysics with experimental collaborations. An intrinsic part of the lab's mission is training a new generation of interdisciplinary researchers with a solid foundation in scientific crafting, metacognition, and rapport.
+This is a new site under construction. It has a repository in GitHub and is generated with GitHub pages. The migration of our [prior lab's website](https://www.luquelab.com) will be completed in August 2022.
diff --git a/docs/knowledge/synthesis.md b/docs/knowledge/synthesis.md
index 4b35852..b5f3576 100644
--- a/docs/knowledge/synthesis.md
+++ b/docs/knowledge/synthesis.md
@@ -5,8 +5,16 @@ nav_order: 1
parent: Knowledge
---
-# The lab's Athena is the synthesis of the current lab's knowledge
+# The Athena initiative
-Narrative and visual synthesis of the scientific knowledge accumulated by the lab over the years.
+## The three foundations of Athena:
+1. Constant restructuring (refactoring) of scienctific knowldege.
+ + Restraining the spontaneous increase of scientific noise (entropy).
+2. Sharing state-of-the-art science transparently and unpretentiously.
+ + Reducing inaccessibility of scientific knowledge and bombastic rhetoric.
+3. Governing scientific knowledge of the people, for the people, and by the people.
+ + Decoupling knowledge from journal publications (validation, advertisement, and promotion).
-Connection with other Athenas and repositories.
+## The lab's Athena is the synthesis of the current lab's knowledge
++ Narrative and visual synthesis of the scientific knowledge accumulated by the lab over the years.
++ Connection with other Athenas and repositories.
diff --git a/docs/organization/Luque_Lab_Events_Record.csv b/docs/organization/Luque_Lab_Events_Record.csv
new file mode 100644
index 0000000..6900233
--- /dev/null
+++ b/docs/organization/Luque_Lab_Events_Record.csv
@@ -0,0 +1,25 @@
+Date,Agenda
+2021-09-02,General Lab Session
+2021-09-09,Brandon Ricafrente's presention
+2021-09-14,Garmann & Luque Viral Assembly Workshop (Brandon Ricafrente's presentation)
+2021-09-16,Diana Lee's presentation (Qualifying Practice)
+2021-09-16,Garmann & Luque Viral Assembly Workshop (Rees Garmann's presentation)
+2021-09-23,Diana Lee's presentation (Qualifying Practice)
+2021-09-24,Diana Lee's Qualifying Exam
+2021-09-30,Colin Brown's presentation
+2021-10-07,Icosahedral Catalogue project
+2021-10-14,Icosahedral Catalogue project
+2021-10-21,Icosahedral Catalogue project
+2021-10-22,Biomath Meeting (Michelle Ann from Segall Lab presentation)
+2021-10-26,Capsid Workshop (White Lab presentation)
+2021-10-27,Capsid Workshop (Luque Lab presentation)
+2021-10-28,Paul Turner's talk on phage therapy
+2021-11-9,Capsid Workshop (White Lab presentation)
+2021-11-11,Capsid Workshop (Luque Lab presentation)
+2021-11-18,Aurora Vogel's presentation
+2021-12-2,Caitlin Bartels' presentation
+2021-12-3,Ugly Sweater Christmas Pub Crawl
+2021-12-6,Capsid Assembly Workshop (Garmann Lab)
+2021-12-8,Capsid Assembly Workshop (Garmann Lab)
+2021-12-9,Brandon Ricafrente's presentation
+2021-12-16,Aurora Vogel's presentation
diff --git a/docs/organization/PVOH_project_description.md b/docs/organization/PVOH_project_description.md
new file mode 100644
index 0000000..87311b6
--- /dev/null
+++ b/docs/organization/PVOH_project_description.md
@@ -0,0 +1,86 @@
+---
+layout: default
+title: Research plan
+nav_order: 1
+parent: Recruiting
+grand_parent: Organization
+has_children: false
+---
+
+# Project: Perpetual Viral Origins
++ ### Principal Investigators: [Forest Rohwer](https://scholar.google.com/citations?hl=en&user=ALEZEdoAAAAJ), [Rob Edwards](https://scholar.google.com/citations?hl=en&user=e7fvl1kAAAAJ), [Anca Segall](https://scholar.google.com/citations?hl=en&user=p1mzR2gAAAAJ), and [Antoni Luque](https://scholar.google.com/citations?user=ytvnI68AAAAJ&hl=en).
++ ### Centers involved: [Viral Information Institute](https://viralization.org) and [Flinders Accelerator for Microbiome Exploration](https://fame.flinders.edu.au).
+
+The research plan is a guide to the intellectual work to be developed in the postdoctoral fellowship. However, as the research unfolds, the priorities and scope of the research may pivot.
+
+## Summary
+This project will combine laboratory, bioinformatics, and computer simulations to compare and test the *perpetual viral origins hypothesis* versus the paradigmatic *spillover hypotheses*. The project is the joint effort of four groups with different expertise and will train three postdoctoral fellows in interdisciplinary science. The research is supported by the Gordon and Betty Moore Foundation.
+
+The *perpetual viral origins hypothesis* predicts that new viruses arise continually from a globally distributed gene pool of *virus-generating elements*. The *spillover hypothesis* is based on descending by modification and predicts that new viruses form as variants of existing viruses. Testing the *perpetual viral origins hypothesis* has major implications for understanding the origin of viruses. Discarding it would imply a limited capacity of viruses to adapt to new environmental conditions. Confirming it would imply that completely new viruses can form regularly and could amplify genes that may help adapt life to dramatic environmental changes. Rates of spillovers and the perpetual origin of new viruses will be quantified and used to parameterize ecological and evolutionary models. Consolidated databases of the global virome will be developed and analyzed to identify novel, unknown proteins, diversity-generating retroelements (DGRs), and *small capsid elements*.
+
+## Research plan associated with the modeling part of the project
+The postdoctoral fellow leading the modeling effort will work under the supervision of Antoni Luque as part of the Viral Information Institute, Computational Science Research Center, and Departments of Mathematics & Statistics at San Diego State University. This part of the project is associated with the third aim of the grant. It will develop the mathematical prediction of biophysical and evolutionary parameters governing the *perpetual viral origins hypothesis*. This aim will determine the minimal structural protein sequence length and composition capable of forming stable capsids that store their own genomic information (Year 1). Examples of such proteins will be searched bioinformatically within small circular contigs (< 2 kbp) obtained from viromes and metagenomes (Year 2). The probability and cost of selecting and propagating adaptive mutations in *virus-generating elements* will be estimated by combining molecular evolutionary theory, the metabolic theory of ecology, and stochastic simulations (Year 3).
+
+**Determine the smallest structural proteins capable of assembling *virus-generating elements*.** In Year 1, the shortest structural protein (amino-acid length) capable of building a stable *virus-generating element* capsid will be determined for helical, tetrahedral, octahedral, dodecahedral, and icosahedral capsid geometries (Luque and Reguera 2010; Luque et al. 2010; Twarock and Luque 2019) (Figure 1a). The selected capsids will have an internal volume that accommodates at least the genomic information of the encoded structural protein. The structural protein’s amino-acid composition (hydrophobic and charged residues) will be estimated using the classical nucleation theory of capsids, which will establish the protein-protein and protein-genome interactions necessary to assemble *virus-generating elements* (VGE) particles spontaneously (Luque et al. 2012). The interaction strength will determine the protein surface (number of amino acids) containing hydrophobic patches (for protein-protein interactions) and positively charged patches (for protein-genome interactions) (Kegel and Schoot Pv 2004). These interaction patches will be selected to produce a capsid assembly rate constrained to occur within-host generation times observed environmentally (Luque and Silveira 2020; Silveira, Luque, and Rohwer 2021). This will determine the assembly yield and decay rate of the different VGE capsid geometries. Coarse-grained molecular simulations will validate and refine the predictions from classical nucleation theory (Luque, Zandi, and Reguera 2010; Luque et al. 2012, 2014; Luque, Ozer, and Schlick 2016), providing the assembly pathway and kinetic traps of the most stable VGE geometries.
+
+
+**Figure 1. Structural self-replicating *virus-generating elements*.** a) The genome capacity of capsids increases with its geometrical complexity (number of capsid protein units). b) Targeted small circular genomes (direct terminal repeats, DTR) from metagenomes and smallest structural candidate found in our prior analysis, including the folded capsid protein (CP) and predicted capsid enclosing the genome.
+
+**Environmental evidence of structural proteins forming *virus-generating elements*.** Small circular contigs will be selected from metagenomes and viromes. The selected contigs will have lengths of smaller than 2 kb, at least one open reading frame, and direct terminal repeats > 20 bp (Figure 1b). This will push the limits of the small putative viral genomes that we have obtained recently (Luque et al. 2020). The proteins will be folded with I-TASSER and AlphaFold. The contig length will determine the possible capsid geometries. The stability of the capsids will be tested using coarse-grained molecular simulations (Luque, Zandi, and Reguera 2010; Luque et al. 2012, 2014; Luque, Ozer, and Schlick 2016). The proteins forming stable capsids will be compared with known (viral and non-viral) shell proteins phylogenetically (MUSCLE) and structurally (DALI). The contigs containing these potential structural *virus-generating element* proteins will be synthesized as part of the experiments in specific aim one.
+
+**Evolutionary consequences of the *perpetual viral origins hypothesis*.** The *spillover hypothesis* predicts that viral evolution largely occurs via mutational drift, whereas the *perpetual viral origins hypothesis* predicts that viral evolution occurs largely via reassortment of genes and/or modules into novel genomic arrangements. In Year 3, a theoretical analysis will estimate the impact of *virus-generating elements* propagating fixed mutations in a community via recombination using the rates observed in specific aim one. The theoretical model will estimate the energetic cost and propagation dynamics of an adapted gene within a microbial community in the *spillover hypothesis* versus the *perpetual viral origins hypothesis*. The model will assume that a community faces a change in environmental conditions (e.g., temperature, salinity) and that a specific mutation in a gene would provide a positive selection. The probability of generating this specific mutation in *virus-generating elements*, viruses, and bacteria in a community will be calculated using Drake’s rule (Drake et al. 1998; Sung et al. 2012). The number of replication cycles required will be coupled with the metabolic theory of ecology to estimate the heat produced by the community to fix that mutation in genomes (Brown et al. 2004; DeLong et al. 2010). A system of seven dynamical equations will monitor the propagation of this fixed mutation and the accumulated heat. It will use three compartments: Bacteria, viruses, and *virus-generating elements*. Each compartment will contain two effective subpopulations: One with and one without the fixed mutation. In the *perpetual viral origins hypothesis*, the mutation could be fixed independently in the viral, bacterial, or *virus-generating elements* compartments, and recombination can propagate the mutation between compartments. In the *spillover hypothesis* model, the *virus-generating elements* compartment will not be included. The system will be parametrized stochastically using Latin hypercube sampling (1,000,000 samples per scenario). The parameters will be determined by combining meta-analyses of empirical data and first-principle physical estimates, analogously to our recent approach investigating the integration and propagation of proviruses in bacterial communities (Joiner et al. 2019; Anthenelli et al. 2020; Luque and Silveira 2020; Silveira, Luque, and Rohwer 2021). The dynamics of the fraction of bacterial subpopulations accumulating the fixed mutation and total dissipated heat (energy cost) will be the main outcomes comparing the *spillover hypothesis* versus the *perpetual viral origins hypothesis*. Our working hypothesis is that *virus-generating elements* will lead to a more punctuated and energetically efficient generation and propagation of fixed mutations. This is based on our initial estimates, which predict that the metabolic cost of fixing a mutation increases exponentially with the genome size.
+
+**Deliverables:**
+1. We will determine the theoretical limit of small structural protein properties (amino-acid length and composition) capable of building self-replicating capsids.
+2. We will identify bioinformatically small capsid *virus generating elements* for comparison with the theoretical limit predicted in deliverable one and test in specific aim one.
+3. We will determine the energetic theoretical cost and dynamic of fixing a mutation in a microbial community via the *perpetual viral origins hypothesis* versus the *spillover hypothesis*.
+
+## References
+Anthenelli, Maxwell, Emily Jasien, Robert Edwards, Barbara Bailey, Ben Felts, Parag Katira, James Nulton, Peter Salamon, Forest Rohwer, Cynthia B. Silveira, Antoni Luque. 2020. “Phage and Bacteria Diversification through a Prophage Acquisition Ratchet.” BioRxiv. .
+
+Brown, James H., James F. Gillooly, Andrew P. Allen, Van M. Savage, and Geoffrey B. West. 2004. “Toward a Metabolic Theory of Ecology.” Ecology 85 (7): 1771–89. .
+
+DeLong, John P., Jordan G. Okie, Melanie E. Moses, Richard M. Sibly, and James H. Brown. 2010. “Shifts in Metabolic Scaling, Production, and Efficiency across Major Evolutionary Transitions of Life.” Proceedings of the National Academy of Sciences of the United States of America 107 (29): 12941–45. .
+
+Drake, J. W., B. Charlesworth, D. Charlesworth, and J. F. Crow. 1998. “Rates of Spontaneous Mutation.” Genetics 148 (4): 1667–86. .
+
+Joiner, Kevin L., Arlette Baljon, Jeremy Barr, Forest Rohwer, and Antoni Luque. 2019. “Impact of Bacteria Motility in the Encounter Rates with Bacteriophage in Mucus.” Scientific Reports 9 (1): 16427. .
+
+Kegel, Willem K., and Paul van der Schoot Pv. 2004. “Competing Hydrophobic and Screened- Coulomb Interactions in Hepatitis B Virus Capsid Assembly.” Biophysical Journal 86 (6): 3905–13. .
+
+Luque, Antoni, and David Reguera. 2010. “The Structure of Elongated Viral Capsids.” Biophysical Journal 98 (12): 2993–3003. .
+
+Luque, Antoni, Roya Zandi, and David Reguera. 2010. “Optimal Architectures of Elongated Viruses.” Proceedings of the National Academy of Sciences of the United States of America 107 (12): 5323–28. .
+
+Luque, Antoni, David Reguera, Alexander Morozov, Joseph Rudnick, and Robijn Bruinsma. 2012. “Physics of Shell Assembly: Line Tension, Hole Implosion, and Closure Catastrophe.” The Journal of Chemical Physics 136 (18): 184507. .
+
+Luque, Antoni, Rosana Collepardo-Guevara, Sergei Grigoryev, and Tamar Schlick. 2014. “Dynamic Condensation of Linker Histone C-Terminal Domain Regulates Chromatin Structure.” Nucleic Acids Research 42 (12): 7553–60. .
+
+Luque, Antoni, Gungor Ozer, and Tamar Schlick. 2016. “Correlation among DNA Linker Length, Linker Histone Concentration, and Histone Tails in Chromatin.” Biophysical Journal 110 (11): 2309–19. .
+
+Luque, Antoni, and Cynthia B. Silveira. 2020. “Quantification of Lysogeny Caused by Phage Coinfections in Microbial Communities from Biophysical Principles.” MSystems 5 (5). .
+
+Luque, Antoni, Sean Benler, Diana Y. Lee, Colin Brown, and Simon White. 2020. “The Missing Tailed Phages: Prediction of Small Capsid Candidates.” Microorganisms 8 (12). .
+
+Silveira, Cynthia B., Antoni Luque, and Forest Rohwer. 2021. “The Landscape of Lysogeny
+across Microbial Community Density, Diversity and Energetics.” Environmental
+Microbiology, June. .
+
+Sung, Way, Matthew S. Ackerman, Samuel F. Miller, Thomas G. Doak, and Michael Lynch. 2012. “Drift-Barrier Hypothesis and Mutation-Rate Evolution.” Proceedings of the
+National Academy of Sciences of the United States of America 109 (45): 18488–92. .
+
+Twarock, Reidun, and Antoni Luque. 2019. “Structural Puzzles in Virology Solved with an
+Overarching Icosahedral Design Principle.” Nature Communications 10 (1): 4414. .
+
+
+
+
+
+
+
+
+
+
+
+
+
diff --git a/docs/organization/agenda.md b/docs/organization/agenda.md
index bee59d1..6d9b726 100644
--- a/docs/organization/agenda.md
+++ b/docs/organization/agenda.md
@@ -7,50 +7,13 @@ parent: Organization
# Lab's Agenda
-### September 1, Thursday: Lab meeting
-+ 2:00 pm - 3:30 pm
-+ General lab session.
+### January 14, Sat: Quarterly Meeting
++ Plan TDB
-### September 9, Thursday: Lab meeting
-+ 9:00 am - 11:30 am
-+ Brandon Ricafrente presenting (20 min).
+### January 21, Fri: SRS Registration Deadline
++ Diana & Colin - talks
++ Brandon & Caitlin - posters
++ Aurora - TDB
-### September 16, Thu: Lab meeting
-+ 2:00 pm - 3:00 pm
-+ Diana Lee presenting (35 min).
+### March 4 & 5, Fri/Sat: SRS Days
-### September 14 & 16, Tue & Thu: Garmann and Luque labs
-+ 3:00 pm - 5:00 pm
-+ GMCS 405
-+ Viral assembly workshop.
-+ Brando Ricafrente presenting on the 14th.
-+ Rees Garmann's group presenting on the 16th.
-+ Bar after? (play by ear).
-
-### September 23, Thu: Lab meeting
-+ 9:00 am - 11:30 am
-+ Caitlin Bartels presenting (20 min).
-
-### September 24, Fri: Diana's qualifying
-+ 2:00 pm – 3:30 pm
-
-### September 30, Thu: Lab meeting
-+ 2:00 pm - 3:30 pm
-+ Colin Brown presenting (20 min).
-
-### October 7, Thu: Lab meeting
-+ 9:00 am - 11:30 am
-+ Aurora Vogel presenting (20 min).
-
-### October 14, Thu: Lab meeting
-+ 2:00 pm - 3:30 pm
-+ Toni Luque presenting (20 min).
-
-### October 21, Thu: Lab meeting
-+ 9:00 am - 11:30 am
-+ General lab session.
-
-### October 26-29, Tue-Fri: White and Luque labs
-+ Time TBD (East coast)
-+ Phage capsid structure workshop.
-+ Agenda TBD on Oct 1.
diff --git a/docs/organization/images/figure_small_capsids_2021-08-25.svg b/docs/organization/images/figure_small_capsids_2021-08-25.svg
new file mode 100644
index 0000000..f2aa153
--- /dev/null
+++ b/docs/organization/images/figure_small_capsids_2021-08-25.svg
@@ -0,0 +1,4113 @@
+
+
+
diff --git a/docs/organization/people.md b/docs/organization/people.md
index 2fd564b..844e75c 100644
--- a/docs/organization/people.md
+++ b/docs/organization/people.md
@@ -32,3 +32,6 @@ has_children: true
## Caitlin Bartels
+ Undergraduate student in Biology.
+
+## Jessica Vogt
++ Undergraduate student in Computer Science.
diff --git a/docs/organization/recruiting.md b/docs/organization/recruiting.md
new file mode 100644
index 0000000..2200754
--- /dev/null
+++ b/docs/organization/recruiting.md
@@ -0,0 +1,19 @@
+---
+layout: default
+title: Recruiting
+nav_order: 6
+parent: Organization
+has_children: true
+---
+
+# Open positions in the lab
+
+## Postdoctoral Position in Viruses & Computational Biophysics in San Diego
+
+The [Viral Information Institute](https://viralization.org) has an open postdoctoral fellowship in computational biophysics funded by the [Gordon and Betty Moore Foundation](https://www.moore.org) to investigate the origin of viruses. The research is at the interface of physical virology, viral ecology, and viral evolution, and it will combine geometrical modeling, molecular simulations, and bioinformatics. The ideal candidate would have experience modeling biological systems and a good foundation in mathematics and physics. In addition, experience in molecular dynamics would be a plus. The fellowship is funded for up to three years.
+
+The postdoctoral fellow will join an interdisciplinary group with four principal investigators: [Forest Rohwer](https://scholar.google.com/citations?hl=en&user=ALEZEdoAAAAJ), [Rob Edwards](https://scholar.google.com/citations?hl=en&user=e7fvl1kAAAAJ), [Anca Segall](https://scholar.google.com/citations?hl=en&user=p1mzR2gAAAAJ), and [Antoni Luque](https://scholar.google.com/citations?user=ytvnI68AAAAJ&hl=en). In addition, the postdoctoral fellow will collaborate with a diverse group of postdocs, graduate students, and undergraduate students as part of the [Viral Information Institute](https://viralization.org) at [San Diego State University](https://www.sdsu.edu).
+
+Please, email Antoni Luque at if you need any additional information. To apply to the fellowship, send an email with the subject "GBMF postdoc candidate" and include a letter of motivation, CV, and three contact references.
+
+The announcement is available online at this [link](https://luquelab.github.io/Athena/organization/recruiting.html), which will reflect any updates.
diff --git a/docs/products/.DS_Store b/docs/products/.DS_Store
new file mode 100644
index 0000000..c66f381
Binary files /dev/null and b/docs/products/.DS_Store differ
diff --git a/docs/products/courses/GitHub.md b/docs/products/courses/GitHub.md
deleted file mode 100644
index c762253..0000000
--- a/docs/products/courses/GitHub.md
+++ /dev/null
@@ -1,12 +0,0 @@
----
-layout: default
-title: GitHub
-nav_order: 2
-parent: Courses
----
-
-# GitHub fundamentals for lab members
-
-+ GitHub repos
-+ Git in command line
-+ GitHub and Jekyll
\ No newline at end of file
diff --git a/docs/products/courses/git_hub_how_to_close_an_issue_2021-09-27.mp4 b/docs/products/courses/git_hub_how_to_close_an_issue_2021-09-27.mp4
new file mode 100644
index 0000000..583fde3
Binary files /dev/null and b/docs/products/courses/git_hub_how_to_close_an_issue_2021-09-27.mp4 differ